Catalog # |
Size |
Price |
|
|---|---|---|---|
| EK-071-68 | 96 wells | $563 |
|
| Recommended | ||||||||
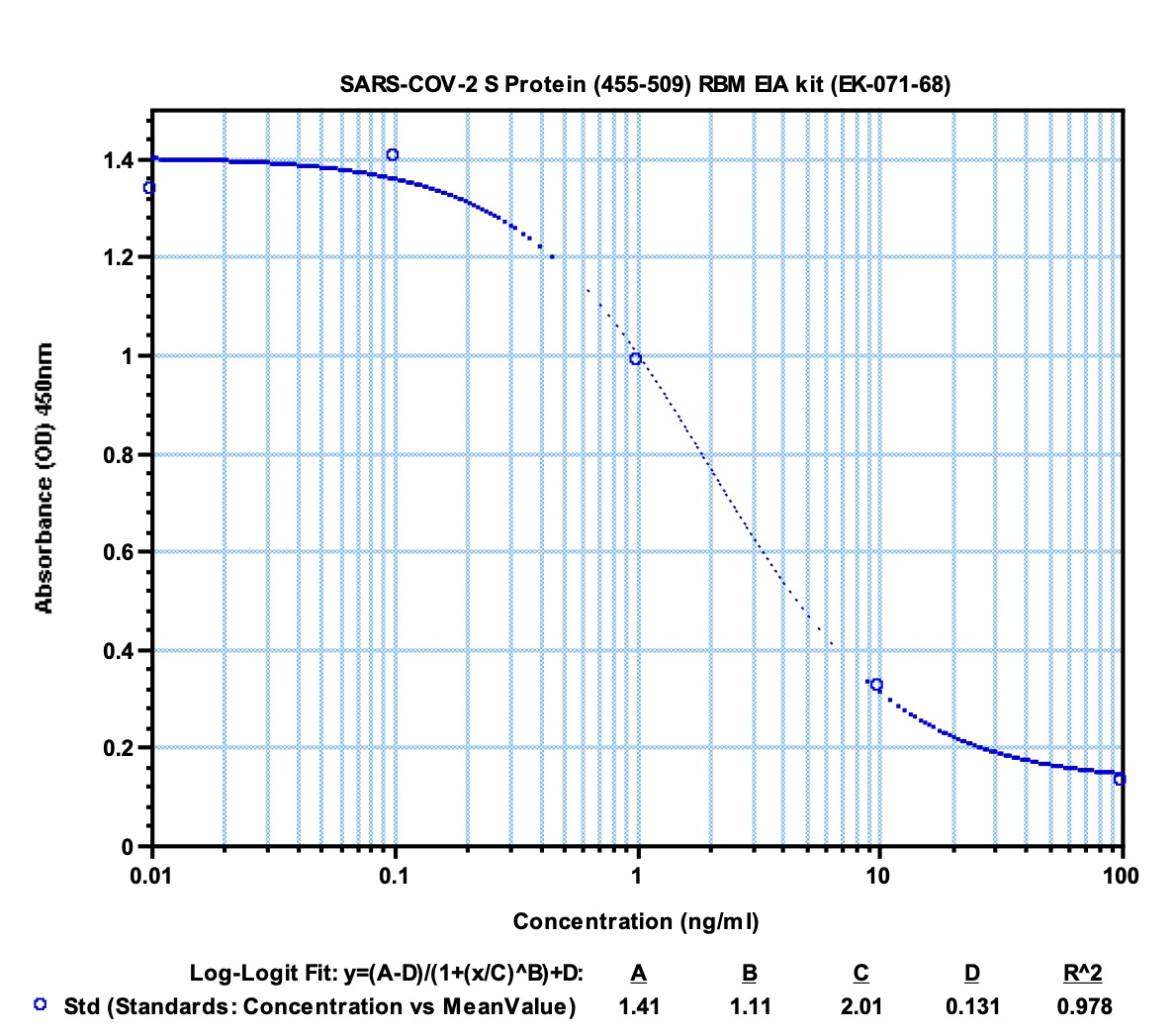
 Data may differ slightly based on lot. | 0.1 ng/ml | ||||||||
|
| 0.64 - 12.6 ng/ml | ||||||||
| |||||||||
|
| <10% | ||||||||
|
| <15% | ||||||||
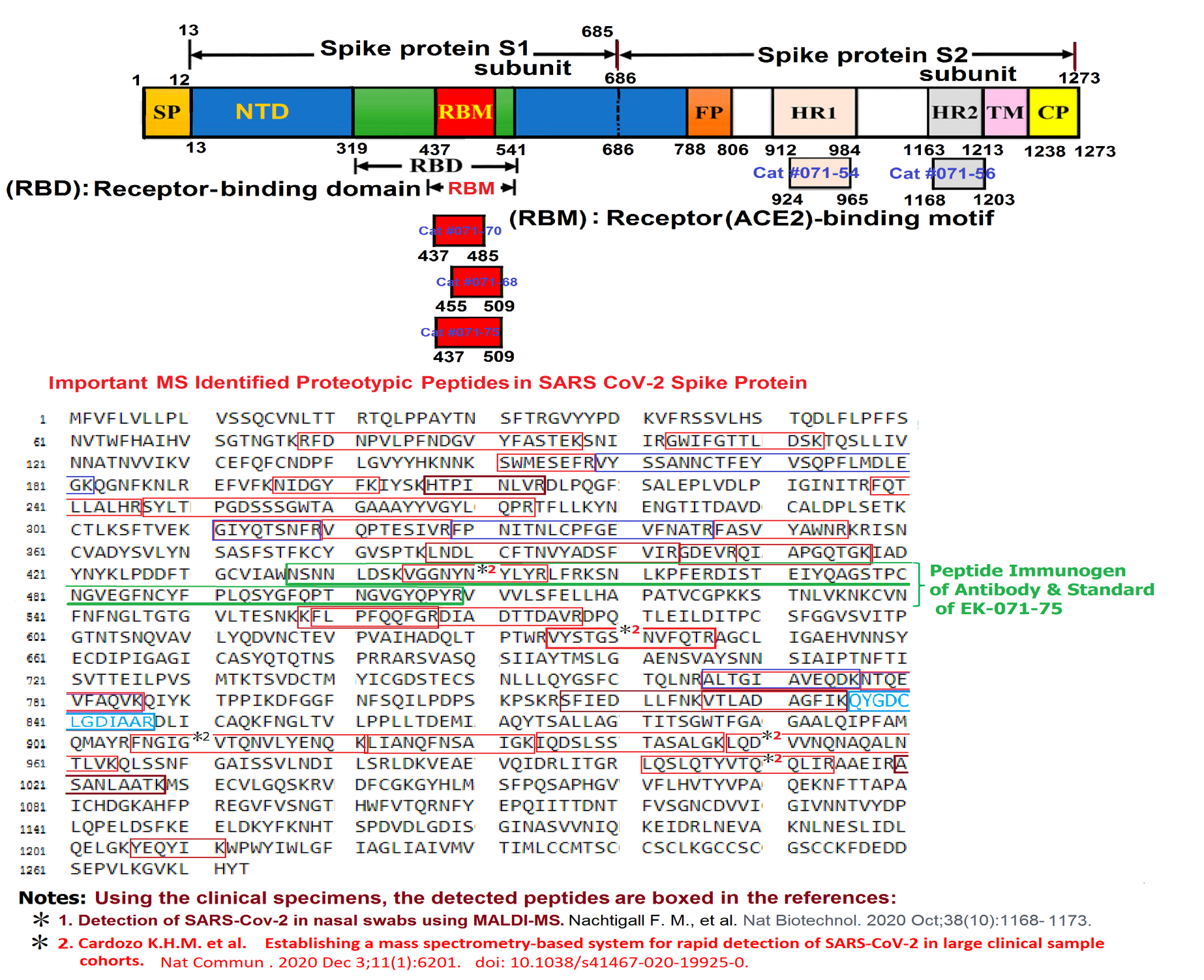

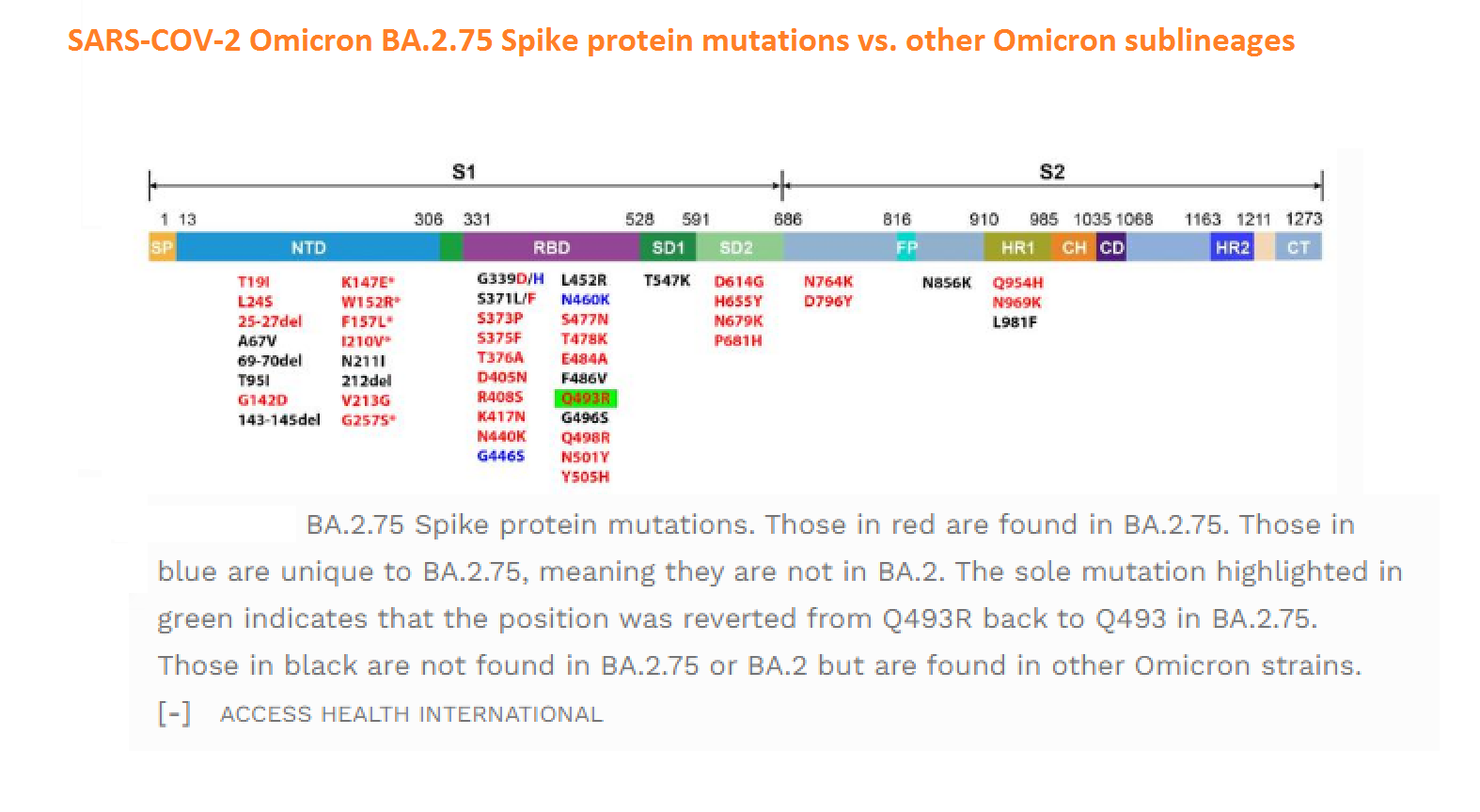
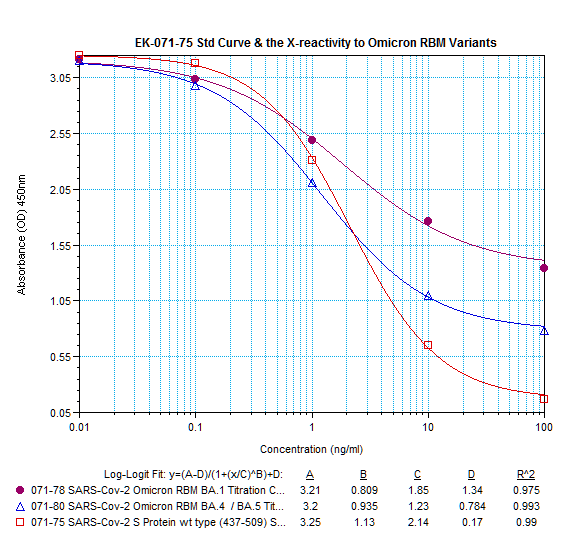

Abstract: Since the end of 2021, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) Omicron variant outcompeted other variants and took over the world. After the emergence of original Omicron BA.1, Omicron BA.2 subvariant emerged and outcompeted BA.1. As of July 2022, some BA.2 subvariants, including BA.2.12.1, BA.4 and BA.5, emerged in multiple countries and begun outcompeting original BA.2. Moreover, a novel BA.2 subvariant, BA.2.75, was detected in eight countries including India at the end of June 2022, and preliminary investigations suggest that BA.2.75 is more transmissible over the other BA.2 subvariants. On July 7, 2022, the WHO classified BA.2.75 as a variant-of-concern lineage under monitoring. We have recently demonstrated that BA.4/5 is highly resistant to a therapeutic monoclonal antibody, cilgavimab, than BA.2. The resistance of SARS-CoV-2 variants to therapeutic antibodies can be attributed to the mutations in the viral spike protein. Compared to the BA.2 spike, BA.2.12.1 and BA.4/5 respectively bear two and four mutations in their spike proteins. On the other hand, the majority of BA.2.75 spike bears nine substitutions. The fact that the mutation number in the BA.2.75 spike is larger than those in the BA.4/5 spike raises the possibility that the BA.2.75 spike significantly reduces sensitivity towards therapeutic monoclonal antibodies than BA.2 and BA.4/5. In this study, we generated pseudoviruses harboring the spike proteins of BA.2.75, BA.4/5 and BA.2 and evaluated the efficacy of ten therapeutic monoclonal antibodies and three antibody cocktails against BA.2.75
Yamasoba D, Kimura I, Kosugi Y, et al. Neutralization Sensitivity of Omicron BA.2.75 to Therapeutic Monoclonal Antibodies. Microbiology; 2022.
Abstract: The Omicron variant of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has now spread throughout the world. We used computational tools to assess the spike infectivity, transmission, and pathogenicity of Omicron (BA.1) and sub-variants (BA.1.1, BA.2, and BA.3) in this study. BA.1 has 39 mutations, BA.1.1 has 40 mutations, BA.2 has 31 mutations, and BA.3 has 34 mutations, with 21 shared mutations between all. We observed 11 common mutations in Omicron's receptor-binding domain (RBD) and sub-variants. In pathogenicity analysis, the Y505H, N786K, T95I, N211I, N856K, and V213R mutations in omicron and sub-variants are predicted to be deleterious. Due to the major effect of the mutations characterizing in the RBD, we found that Omicron and sub-variants had a higher positive electrostatic surface potential. This could increase interaction between RBD and negative electrostatic surface potential human angiotensin-converting enzyme 2 (hACE2). Omicron and sub-variants had a higher affinity for hACE2 and the potential for increased transmission when compared to the wild-type (WT). Negative electrostatic potential of N-terminal domain (NTD) of the spike protein value indicates that the Omicron variant binds receptors less efficiently than the WT. Given that at least one receptor is highly expressed in lung and bronchial cells, the electrostatic potential of NTD negative value could be one of the factors contributing to why the Omicron variant is thought to be less harmful to the lower respiratory tract. Among Omicron sub-lineages, BA.2 and BA.3 have a higher transmission potential than BA.1 and BA.1.1. We predicted that mutated residues in BA.1.1 (K478), BA.2 (R400, R490, and R495), and BA.3 (R397 and H499) formation of new salt bridges and hydrogen bonds. Omicron and sub-variant mutations at Receptor-binding Motif (RBM) residues such as Q493R, N501Y, Q498, T478K, and Y505H all contribute significantly to binding affinity with human ACE2. Interactions with Omicron variant mutations at residues 493, 496, 498, and 501 seem to restore ACE2 binding effectiveness lost due to other mutations like K417N.
Omicron... and sub-variants...: A comparative sequence and structural-based computational assessment
Abstract: Whole-genome sequencing (WGS) is the gold standard for characterizing the severe acute respiratory syndrome coronavirus 2(SARS-CoV-2) genome and identification of new variants. However, the cost involved and time needed for WGS prevent routine, rapid clinical use. This study aimed to develop a quick and cost-effective surveillance strategy for SARS-CoV-2 variants in saliva and nasal swab samples by spike protein receptor-binding-motif (RBM)-targeted Sanger sequencing. Saliva and nasal swabs prescreened for the presence of the nucleocapsid (N) gene of SARS-CoV-2 were subjected to RBM-specific single-amplicon generation and Sanger sequencing. Sequences were aligned by CLC Sequence Viewer 8, and variants were identified based upon specific mutation signature. Based on this strategy, the present study identified Alpha, Beta/Gamma, Delta, and Omicron variants in a quick and cost-effective manner. IMPORTANCE The coronavirus disease 2019 (COVID-19) pandemic resulted in 427 million infections and 5.9 million deaths globally as of 21 February 2022. SARS-CoV-2, the causative agent of the COVID-19 pandemic, frequently mutates and has developed into variants of major public health concerns. Following the Alpha variant (B.1.1.7) infection wave, the Delta variant (B.1.617.2) became prevalent, and now the recently identified Omicron (B.1.1.529) variant is spreading rapidly and forming BA.1, BA.1.1, BA.2, BA.3, BA.4, and BA.5 lineages of concern. Prompt identification of mutational changes in SARS-CoV-2 variants is challenging but critical to managing the disease spread and vaccine/therapeutic modifications. Considering the cost involved and resource limitation of WGS globally, an RBM-targeted Sanger sequencing strategy is adopted in this study for quick molecular surveillance of SARS-CoV-2 variants.
Chaki SP, Kahl-McDonagh MM, Neuman BW, Zuelke KA. Receptor-binding-motif-targeted sanger sequencing: a quick and cost-effective strategy for molecular surveillance of sars-cov-2 variants. Narayanasamy P, ed. Microbiol Spectr.
No References
Social Network Confirmation