Abstract: Aberrant activation of Wnt/β-catenin signaling has been associated with the onset and progression of many types of tumors and thus β-catenin represents one attractive intracellular target for cancer therapy. Based on the Axin-derived peptide that binds to β-catenin, two stapled peptides SAHPA1 and xStAx were reported to enhance or impair Wnt/β-catenin signaling, respectively. In this study, we designed PROTACs (proteolysis targeting chimeras) by coupling SAHPA1 or xStAx with the VHL ligand to achieve efficient β-catenin degradation. The obtained xStAx-VHLL sustained β-catenin degradation and manifested strong inhibition of Wnt signaling in cancer cells and in APC -/- organoids. Furthermore, xStAx-VHLL could effectively restrain tumor formation in BALB/C nude mice, and diminish the existing tumors in APC min/+ mice. More importantly, xStAx-VHLL could potently inhibit the survival of colorectal cancer patient-derived organoids. These findings suggest that xStAx-VHLL exhibits the ability of cancer prevention and cure, highlighting the potential of β-catenin degrader PROTACs as a new class of promising anticancer agent.
Liao H, Li X, Zhao L, et al. A PROTAC peptide induces durable β-catenin degradation and suppresses Wnt-dependent intestinal cancer. Cell Discov. 2020;6(1):35.
Abstract: Cancers remain a threat to human health due to the lack of effective therapeutic strategies. Great effort has been devoted to the discovery of drug targets to treat cancers, but novel oncoproteins still need to be unveiled for efficient therapy.Methods: We show that CREPT is highly expressed in pancreatic cancer and is associated with poor disease-free survival. CREPT overexpression promotes but CREPT deletion blocks colony formation and proliferation of pancreatic cancer cells. To provide a proof of concept for CREPT as a new target for the inhibition of pancreatic cancer, we designed a cell-permeable peptide-based proteolysis targeting chimera (PROTAC), named PRTC, based on the homodimerized leucine-zipper-like motif in the C-terminus domain of CREPT to induce its degradation in vivo. Results: PRTC has high affinity for CREPT, with Kd = 0.34 +/- 0.11 μM and is able to permeate into cells because of the attached membrane-transportable peptide RRRRK. PRTC effectively induces CREPT degradation in a proteasome-dependent manner. Intriguingly, PRTC inhibits colony formation, cell proliferation, and motility in pancreatic cancer cells and ultimately impairs xenograft tumor growth, comparable to the effect of CREPT deletion. Conclusions: PRTC-induced degradation of CREPT leads to inhibition of tumor growth, which is promising for the development of new drugs against pancreatic cancer. In addition, using an interacting motif based on the dimerized structure of proteins may be a new way to design a PROTAC aiming at degrading any protein without known interacting small molecules or peptides.
Ma D, Zou Y, Chu Y, et al. A cell-permeable peptide-based PROTAC against the oncoprotein CREPT proficiently inhibits pancreatic cancer. Theranostics. 2020;10(8):3708-3721.
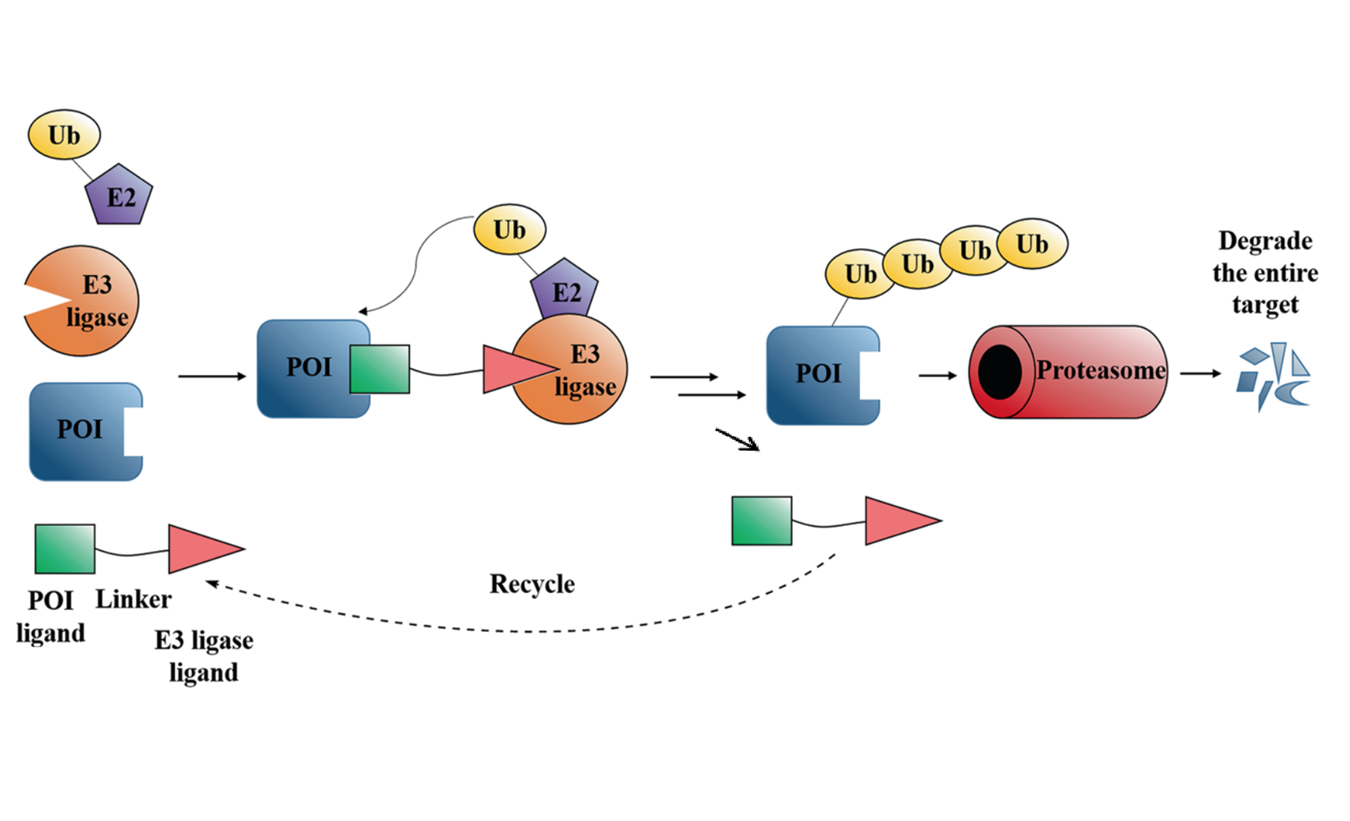
Abstract Targeted protein degradation by small-molecule degraders represents an emerging mode of action in drug discovery. Proteolysis targeting chimeras (PROTACs) are small molecules that can recruit an E3 ligase and a protein of interest (POI) into proximity, leading to induced ubiquitination and degradation of the POI by the proteasome system. To date, the design and optimization of PROTACs remain empirical due to the complicated mechanism of induced protein degradation. Nevertheless, it is increasingly appreciated that profiling step-by-step along the ubiquitin-proteasome degradation pathway using biochemical and biophysical assays are essential in understanding the structure-activity relationship and facilitating the rational design of PROTACs. This review aims to summarize these assays and to discuss the potential of expanding the toolbox with other new techniques.
Liu X, Zhang X, Lv D, Yuan Y, Zheng G, Zhou D. Assays and technologies for developing proteolysis targeting chimera degraders. Future Medicinal Chemistry. Published online May 20, 2020:fmc-2020-0073.
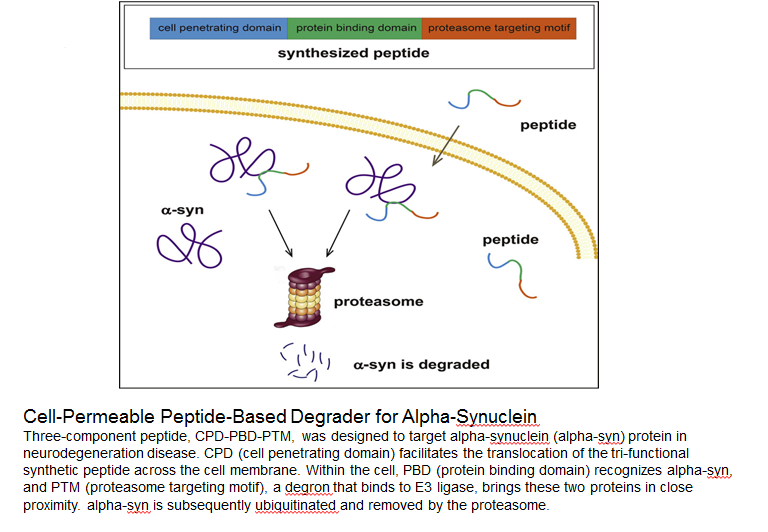
Abstract: α-Synuclein (α-syn) overload is strongly associated with Parkinson disease (PD), and reduction of the α-syn level by targeting the peptide-based system through the autophagy-lysosomal pathway (ALP) is a promising strategy to delay PD progression. However, if the ALP is comprised, targeting the peptide-based proteasomal degradation system would be a good alternative. In this study, we designed a fusion peptide containing an α-syn-binding domain and a short strong proteasome-targeting motif. Our results reveal that this peptide could specifically bind to α-syn, and direct it to the proteasomes for degradation in a recombinant expression system. Furthermore, by adding a membrane-penetrating motif to this fusion peptide, we demonstrated that it could penetrate into cells and consequently suppress the cellular α-syn level through proteasome degradation in a dose- and time-dependent manner. Functionally, these effects rescued the mitochondrial dysfunction and cellular defects caused by α-syn overexpression in the cultured cells and primary neurons.
Qu J, Ren X, Xue F, et al. Specific knockdown of α-synuclein by peptide-directed proteasome degradation rescued its associated neurotoxicity. Cell Chemical Biology. 2020;27(6):751-762.e4.
Introduction: Proteolysis – targeting chimeras (PROTACs) have emerged as a new modality with the potential to revolutionize drug discovery. PROTACs are heterobifunctional molecules comprising of a ligand targeting a protein of interest, a ligand targeting an E3 ligase and a connecting linker. The aim is instead of inhibiting the target to induce its proteasomal degradation.Areas covered: PROTACs, due to their bifunctional design, possess properties that differentiate them from classical inhibitors. A structural analysis, based on published crystal aspects, kinetic features and aspects of selectivity are discussed. Specific types such as homoPROTACs, PROTACs targeting Tau protein and the first PROTACs recently entering clinical trials are examined.Expert opinion: PROTACs have shown remarkable biological responses in challenging targets, including an unprecedented selectivity over protein family members and even efficacy starting from weak or unspecific binders. Moreover, PROTACs are standing out from classical pharmacology by inducing the degradation of the target protein and not merely its inhibition. However, there are also challenges in the field, such as the rational structure optimization, the evolution of computational tools, limited structural data and the greatly anticipated clinical data. Despite the remaining hurdles, PROTACs are expected to soon become a new therapeutic category of drugs.
Konstantinidou M, Li J, Zhang B, et al. PROTACs– a game-changing technology. Expert Opinion on Drug Discovery. 2019;14(12):1255-1268
Although many kinds of therapies are applied in the clinic, drug-resistance is a major and unavoidable problem. Another disturbing statistic is the limited number of drug targets, which are presently only 20-25% of all protein targets that are currently being studied. Moreover, the focus of current explorations of targets are their enzymatic functions, which ignores the functions from their scaffold moiety. As a promising and appealing technology, PROteolysis Targeting Chimeras (PROTACs) have attracted great attention both from academia and industry for finding available approaches to solve the above problems. PROTACs regulate protein function by degrading target proteins instead of inhibiting them, providing more sensitivity to drug-resistant targets and a greater chance to affect the nonenzymatic functions. PROTACs have been proven to show better selectivity compared to classic inhibitors. PROTACs can be described as a chemical knockdown approach with rapidity and reversibility, which presents new and different biology compared to other gene editing tools by avoiding misinterpretations that arise from potential genetic compensation and/or spontaneous mutations. PRTOACs have been widely explored throughout the world and have outperformed not only in cancer diseases, but also in immune disorders, viral infections and neurodegenerative diseases. Although PROTACs present a very promising and powerful approach for crossing the hurdles of present drug discovery and tool development in biology, more efforts are needed to gain to get deeper insight into the efficacy and safety of PROTACs in the clinic. More target binders and more E3 ligases applicable for developing PROTACs are waiting for exploration.
Sun X, Gao H, Yang Y, et al. PROTACs: great opportunities for academia and industry. Signal Transduct Target Ther. 2019;4:64.
Abstract: Induced protein degradation by PROTACs has emerged as a promising strategy to target nonenzymatic proteins inside the cell. The aim of this study was to identify Keap1, a substrate adaptor protein for ubiquitin E3 ligase involved in oxidative stress regulation, as a novel candidate for PROTACs that can be applied in the degradation of the nonenzymatic protein Tau. A peptide PROTAC by recruiting Keap1-Cul3 ubiquitin E3 ligase was developed and applied in the degradation of intracellular Tau. Peptide 1 showed strong in vitro binding with Keap1 and Tau. With proper cell permeability, peptide 1 was found to colocalize with cellular Keap1 and resulted in the coimmunoprecipitation of Tau and Keap1. The results of flow cytometry and western blotting assays showed that peptide 1 can downregulate the intracellular Tau level in both time- and concentration-dependent manner. The application of Keap1 siRNA silencing and the proteasome inhibitor MG132 confirmed that peptide 1 could promote the Keap1-dependent poly-ubiquitination and proteasome-dependent degradation of Tau. The results suggested that using PROTACs to recruit Keap1 to induce the degradation of Tau may show promising character in the treatment of neurodegenerative disease. Besides, our research demonstrated that Keap1 should be a promising E3 ligase adaptor to be used in the design of novel PROTACs.
Lu M, Liu T, Jiao Q, et al. Discovery of a Keap1-dependent peptide PROTAC to knockdown Tau by ubiquitination-proteasome degradation pathway. European Journal of Medicinal Chemistry. 2018;146:251-259.
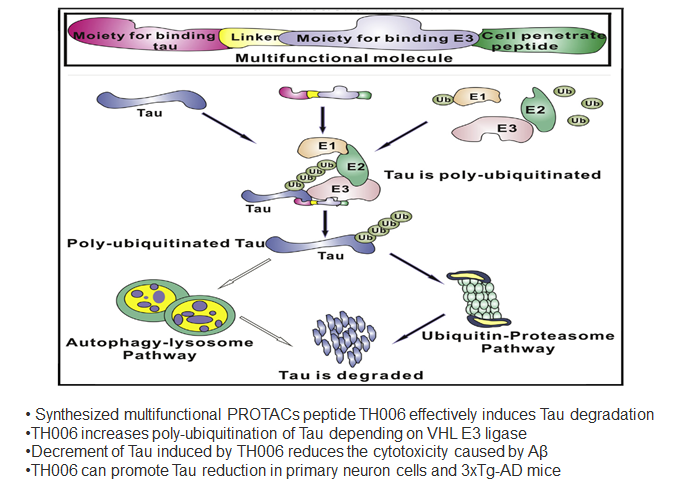
Abstract: Tau, an important pathological protein of Alzheimer's disease (AD), can mediate the toxicity of amyloid β (Aβ). Thus, reduction of Tau with chemical molecules may offer a novel strategy for treating AD. Here, we designed and synthesized a series of multifunctional molecules that contained Tau-recognition moieties and E3 ligase-binding moieties to enhance Tau degradation. Among these molecules, TH006 had the highest activity of inducing Tau degradation by increasing its poly-ubiquitination. The decrement in Tau induced by TH006 could decrease the cytotoxicity caused by Aβ. Furthermore, TH006 could regulate the Tau level in the brain of an AD mouse model. Therefore, partial reduction of Tau with such multifunctional peptides may open up a novel therapeutic strategy for AD treatment.Chu T-T, Gao N, Li Q-Q, et al. Specific knockdown of endogenous tau protein by peptide-directed ubiquitin-proteasome degradation. Cell Chemical Biology. 2016;23(4):453-461.
| Catalog# | Product | Standard Size | Price |
|---|---|---|---|
| 068-96 | PRTC / CREPT degrader | 500ug | $1186 |
| 068-97 | TH006 - Tau degrader | 500ug | $1186 |
| 068-94 | alpha-synuclein degrader | 100ug | $249 |
| 068-95 | xStAx-VHLL / beta-catenin degrader | 500ug | $1186 |
Social Network Confirmation