Abstract: Aggregated insoluble tau is one of two defining features of Alzheimer's disease. Because clinical symptoms are strongly correlated with tau aggregates, drug development and clinical diagnosis need cost-effective and accessible specific fluid biomarkers of tau aggregates; however, recent studies suggest that the fluid biomarkers currently available cannot specifically track tau aggregates. We show that the microtubule-binding region (MTBR) of tau containing the residue 243 (MTBR-tau243) is a new cerebrospinal fluid (CSF) biomarker specific for insoluble tau aggregates and compared it to multiple other phosphorylated tau measures (p-tau181, p-tau205, p-tau217 and p-tau231) in two independent cohorts (BioFINDER-2, n = 448; and Knight Alzheimer Disease Research Center, n = 219). MTBR-tau243 was most strongly associated with tau-positron emission tomography (PET) and cognition, whereas showing the lowest association with amyloid-PET.
Horie K, Salvadó G, Barthélemy NR, et al. CSF MTBR-tau243 is a specific biomarker of tau tangle pathology in Alzheimer’s disease. Nat Med. Published online July 13, 2023.
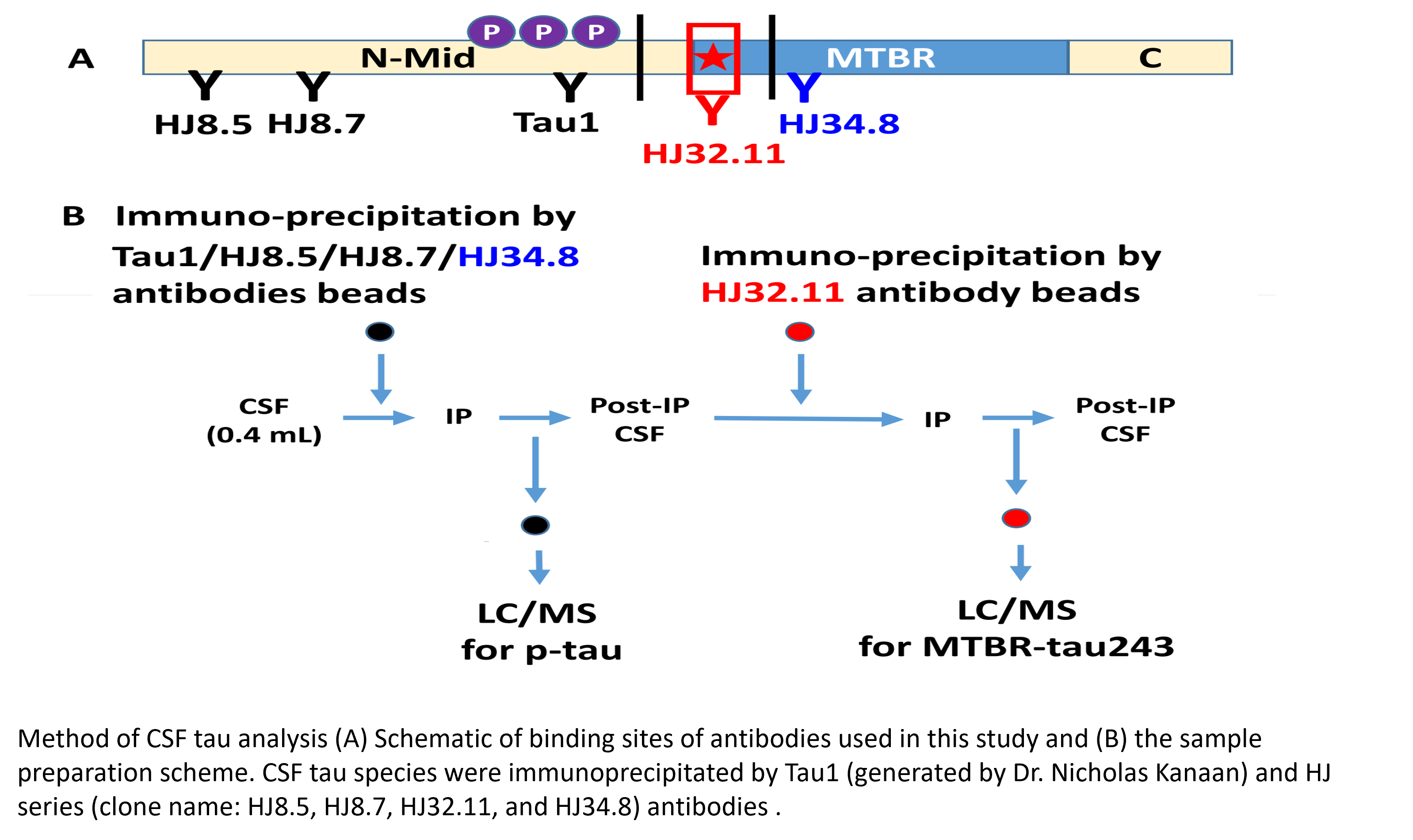
Abstract: Tau is a microtubule associated protein in the brain that aggregates in Alzheimer's disease to form pathological tangles and neurites. Insoluble tau aggregates composed of the microtubule binding region (MTBR) of tau are highly associated with the cognitive and clinical symptoms of Alzheimer's disease. In contrast, levels of soluble forms of tau, such as CSF total tau and phosphorylated tau-181 and tau-217, increase prior to tau aggregation in Alzheimer's disease, but these biomarkers do not measure the MTBR of tau. Thus, how CSF MTBR-tau is altered in Alzheimer's disease remains unclear. In this study, we used sequential immunoprecipitation and chemical extraction methods followed by mass spectrometry to analyse MTBR-tau species in Alzheimer's disease and control CSF. We quantified MTBR-tau-specific regions in the CSF and identified that species containing the region beginning at residue 243 were the most highly correlated with tau PET and cognitive measures. This finding suggests that CSF level of tau species containing the upstream region of MTBR may reflect changes in tau pathology that occur in Alzheimer's disease and could serve as biomarkers to stage Alzheimer's disease and track the development of tau-directed therapeutics.
Horie K, Barthélemy NR, Sato C, Bateman RJ. CSF tau microtubule binding region identifies tau tangle and clinical stages of Alzheimer’s disease. Brain. 2021;144(2):515-527.
Tauopathies feature progressive accumulation of tau amyloids. Pathology may begin when these amplify from a protein template, or seed, whose structure is unknown. We have purified and characterized distinct forms of tau monomer-inert (Mi) and seed-competent (Ms). Recombinant Ms triggered intracellular tau aggregation, induced tau fibrillization in vitro, and self-assembled. Ms from Alzheimer's disease also seeded aggregation and self-assembled in vitro to form seed-competent multimers. We used crosslinking with mass spectrometry to probe structural differences in Mi vs. Ms. Crosslinks informed models of local peptide structure within the repeat domain which suggests relative inaccessibility of residues that drive aggregation (VQIINK/VQIVYK) in Mi, and exposure in Ms. Limited proteolysis supported this idea. Although tau monomer has been considered to be natively unstructured, our findings belie this assumption and suggest that initiation of pathological aggregation could begin with conversion of tau monomer from an inert to a seed-competent form.
Mirbaha H, Chen D, Morazova OA, et al. Inert and seed-competent tau monomers suggest structural origins of aggregation. Elife. 2018;7
Aggregated tau protein is associated with over 20 neurological disorders, which include Alzheimer's disease. Previous work has shown that tau's sequence segments VQIINK and VQIVYK drive its aggregation, but inhibitors based on the structure of the VQIVYK segment only partially inhibit full-length tau aggregation and are ineffective at inhibiting seeding by full-length fibrils. Here we show that the VQIINK segment is the more powerful driver of tau aggregation. Two structures of this segment determined by the cryo-electron microscopy method micro-electron diffraction explain its dominant influence on tau aggregation. Of practical significance, the structures lead to the design of inhibitors that not only inhibit tau aggregation but also inhibit the ability of exogenous full-length tau fibrils to seed intracellular tau in HEK293 biosensor cells into amyloid. We also raise the possibility that the two VQIINK structures represent amyloid polymorphs of tau that may account for a subset of prion-like strains of tau.
Seidler PM, Boyer DR, Rodriguez JA, et al. Structure-based inhibitors of tau aggregation. Nat Chem. 2018;10(2):170-176.
BACKGROUND: Ideally, disease modifying therapies for Alzheimer disease (AD) will be applied during the 'preclinical' stage (pathology present with cognition intact) before severe neuronal damage occurs, or upon recognizing very mild cognitive impairment. Developing and judiciously administering such therapies will require biomarker panels to identify early AD pathology, classify disease stage, monitor pathological progression, and predict cognitive decline. To discover such biomarkers, we measured AD-associated changes in the cerebrospinal fluid (CSF) proteome.
METHODS AND FINDINGS: CSF samples from individuals with mild AD (Clinical Dementia Rating [CDR] 1) (n = 24) and cognitively normal controls (CDR 0) (n = 24) were subjected to two-dimensional difference-in-gel electrophoresis. Within 119 differentially-abundant gel features, mass spectrometry (LC-MS/MS) identified 47 proteins. For validation, eleven proteins were re-evaluated by enzyme-linked immunosorbent assays (ELISA). Six of these assays (NrCAM, YKL-40, chromogranin A, carnosinase I, transthyretin, cystatin C) distinguished CDR 1 and CDR 0 groups and were subsequently applied (with tau, p-tau181 and Aβ42 ELISAs) to a larger independent cohort (n = 292) that included individuals with very mild dementia (CDR 0.5). Receiver-operating characteristic curve analyses using stepwise logistic regression yielded optimal biomarker combinations to distinguish CDR 0 from CDR>0 (tau, YKL-40, NrCAM) and CDR 1 from CDR<1 (tau, chromogranin A, carnosinase I) with areas under the curve of 0.90 (0.85-0.94 95% confidence interval [CI]) and 0.88 (0.81-0.94 CI), respectively.CONCLUSIONS: Four novel CSF biomarkers for AD (NrCAM, YKL-40, chromogranin A, carnosinase I) can improve the diagnostic accuracy of Aβ42 and tau. Together, these six markers describe six clinicopathological stages from cognitive normalcy to mild dementia, including stages defined by increased risk of cognitive decline. Such a panel might improve clinical trial efficiency by guiding subject enrollment and monitoring disease progression. Further studies will be required to validate this panel and evaluate its potential for distinguishing AD from other dementing conditions.
Perrin RJ, Craig-schapiro R, Malone JP, et al. Identification and validation of novel cerebrospinal fluid biomarkers for staging early Alzheimer's disease. PLoS ONE. 2011;6(1):e16032.


| Catalog# | Product | Standard Size | Price |
|---|---|---|---|
| 018-38 | AcPHF6 / Ac-Tau (623-628) | 200 µg | $197 |
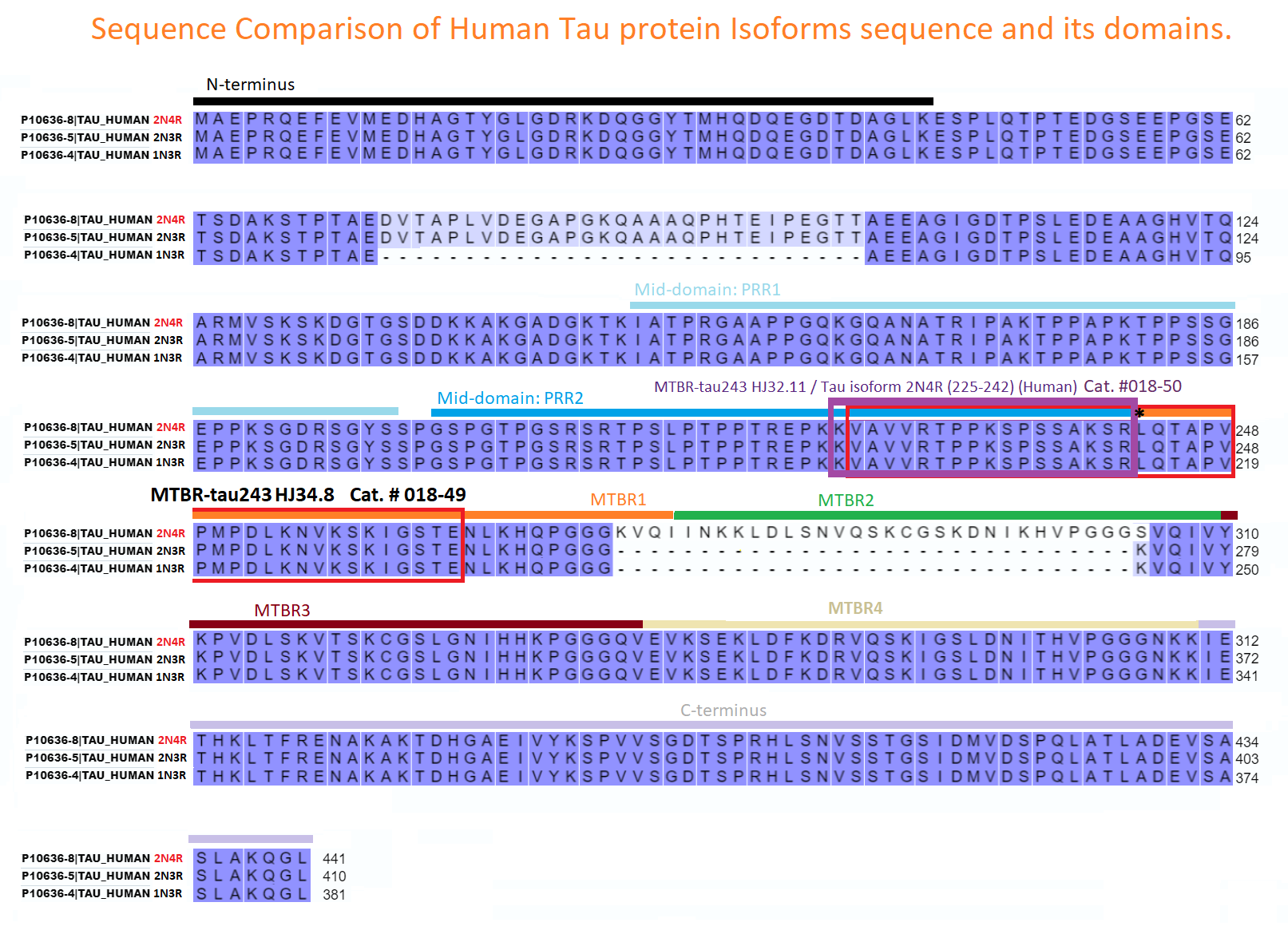
| 018-50 | MTBR-tau243 HJ32.11 / Tau isoform 2N4R (225-242) (Human) | 100 μg | $226 |
| 018-49 | MTBR-tau243 HJ34.8 / Tau isoform 2N4R (226-264) (Human) | 100 μg | $415 |
| 018-87 | Microtubule Associated Protein Tau (591-600) (Human) | 200 µg | $172 |
| 018-86 | Microtubule Associated Protein Tau (592-607) (Human) | 200 µg | $195 |
| 058-70 | Microtubule Associated Protein Tau, C-terminal fragment | 200 µg | $167 |
| SIL-018-49 | [Ala(13C)2 - MTBR-tau243 HJ34.8 / Tau isoform 2N4R (226 -264) (Human) | 20 μg | $1121 |
| SIL-018-50 | [Ala(13C)3 - MTBR-tau243 HJ32.11 / Tau isoform 2N4R (225-242) (Human) | 20 μg | $1121 |



Social Network Confirmation