Catalog # |
Size |
Price |
|
|---|---|---|---|
| FEK-002-01 | 96 wells | $624 |
| View/Download (PDF) - for reference only | |||||||||||||||||||||||||
|
| Suggested Procedure | ||||||||||||||||||||||||
 Measured values represent approximate 'normal' levels in sample, but do not signify the only possible sample levels. Various factors (e.g. sample storage, assay procedure) can affect results. | Human Plasma (extracted): 83.8 pg/ml | ||||||||||||||||||||||||
|
| Recommended | ||||||||||||||||||||||||
|
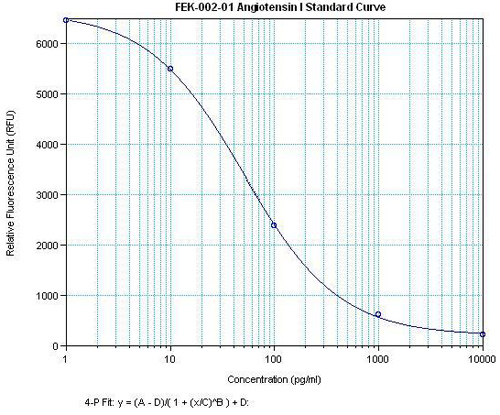
Data may differ slightly based on lot. | 9.6 pg/ml | ||||||||||||||||||||||||
|
Data may differ slightly based on lot. | 9.6 - 279 pg/ml | ||||||||||||||||||||||||
| |||||||||||||||||||||||||
|
| <10% | ||||||||||||||||||||||||
|
| <15% | ||||||||||||||||||||||||
|
| 325 nm | ||||||||||||||||||||||||
|
| 420 nm | ||||||||||||||||||||||||




Background and purpose: Overfeeding increases adipose tissue mass and leptin production and up?regulates the renin?angiotensin system in adipose tissue in rodents. Here, we determined the effect of chronic treatment with the renin inhibitor, aliskiren, in a model of diet?induced obesity in mice, on: (i) body weight, adipose tissue weight and plasma leptin; (ii) food intake and caloric efficiency; and (iii) angiotensin II (Ang II) in adipose tissue.
Experimental approach: Four?week?old C57BL/6J mice (n= 40) received aliskiren (50 mg·kg−1·day−1; 6 weeks) by means of a subcutaneous osmotic Alzet minipump. Animals were given either a low?fat (10% kcal from fat) or a high?fat diet (45% kcal from fat) during this period. Food?intake and body?weight variation were monitored during treatment.
Key results: In addition to a decrease of plasma renin activity, aliskiren reduced body?weight gain, adipose pads and plasma leptin concentration, independent of the diet. In adipose tissue, local concentrations of Ang II were also reduced by aliskiren.
Conclusions and implications: Aliskiren limited the gain of adiposity in young mice. This effect was not due to changes in food intake or caloric efficiency and might be related to a down?regulation of the local renin?angiotensin system in adipose tissue. These effects were accompanied by reduced plasma leptin levels. As Ang II favours differentiation of adipocytes, it is possible that the decreased adipose tissue was linked to changes in adipocyte size and number.
Stucchi P, Cano V, Ruiz-gayo M, Fernández-alfonso MS. Br J Pharmacol. 2009;158(3):771-8.
High levels (μM) of beta amyloid (Aβ) oligomers are known to trigger neurotoxic effects, leading to synaptic impairment, behavioral deficits and apoptotic cell death. The hydrophobic C-terminal domain of Aβ, together with sequences critical for oligomer formation, is essential for this neurotoxicity. However, Aβ at low levels (pM-nM) has been shown to function as a positive neuromodulator and this activity resides in the hydrophilic N-terminal domain of Aβ. An N-terminal Aβ fragment (1-15/16), found in cerebrospinal fluid, was also shown to be a highly active neuromodulator and to reverse Aβ-induced impairments of long-term potentiation. Here, we show the impact of this N-terminal Aβ fragment and a shorter hexapeptide core sequence in the Aβ fragment (Aβcore: 10-15) to protect or reverse Aβ-induced neuronal toxicity, fear memory deficits and apoptotic death. The neuroprotective effects of the N-terminal Aβ fragment and Aβcore on Aβ-induced changes in mitochondrial function, oxidative stress and apoptotic neuronal death were demonstrated via mitochondrial membrane potential, live reactive oxygen species, DNA fragmentation and cell survival assays using a model neuroblastoma cell line (differentiated NG108-15) and mouse hippocampal neuron cultures. The protective action of the N-terminal Aβ fragment and Aβcore against spatial memory processing deficits in APP/PSEN1 (5XFAD) mice was demonstrated in contextual fear conditioning. Stabilized derivatives of the N-terminal Aβcore were also shown to be fully protective against Aβ-triggered oxidative stress. Together, these findings indicate an endogenous neuroprotective role for the N-terminal Aβ fragment, while active stabilized N-terminal Aβcore derivatives offer the potential for therapeutic application.
The renin-angiotensin system (RAS) is a unique hormonal cascade which is composed by multiple enzymes and effector peptides. Recently, new peptides presenting biological activity have been discovered, increasing the complexity of the RAS Here, we evaluated the effects of small peptides of the RAS in coronary bed of rats. Firstly, we examined the direct effect of small angiotensinergic peptides Angiotensin Ang) -(1-5), Ang-(1-4) Ang-(1-3), and Ang-(1-2)] in coronary vessels. Noteworthy, it was observed that Ang-(1-4), Ang-(1-3), and Ang-(1-2) caused a significant reduction in pressure perfusion. Because Ang-(1-2) was the smallest peptide tested and presented the major effect, we decided to investigate its mechanisms of action. The effect of Ang-(1-2) was partially dependent on the Mas receptor, nitric oxide release and angiotensin-converting enzyme. Importantly, Ang-(1-2) reduced the blood pressure of Wistar rats and SHR Interestingly, SHR presented a more pronounced decrease in blood pressure levels than Wistar rats. Altogether, these data showed that angiotensinergic small peptides hold biological activities in coronary bed of rats.
Moraes PL, Kangussu LM, da Silva LG, et al., JrPhysiol Rep. 2017 Nov;5(22). pii: e13505. doi: 10.14814/phy2.13505.
Angiotensin II (Ang II) is a natural mammalian hormone that has been described to exhibit antiplasmodial activity therefore constituting a promising alternative for the treatment of malaria. Despite its promise, the development of Ang II as an antimalarial is limited by its potent induction of vasoconstriction and its rapid degradation within minutes. Here, we used peptide design to perform targeted chemical modifications to Ang II to generate conformationally restricted (disulfide-crosslinked) peptide derivatives with suppressed vasoconstrictor activity and increased stability. Designed constrained peptides were synthesized chemically and then tested for antiplasmodial activity. Two lead constrained peptides were identified (i.e., peptides 1 and 2), each composed of 10 amino acid residues. These peptides exhibited very promising activity in both our Plasmodium gallinaceum (>80%) and Plasmodium falciparum (>40%) models, an activity that was equivalent to that of Ang II, and led to complete suppression of vasoconstriction. In addition, peptide 5 exhibited partial activity towards the pre-erythrocytic stage (98% of activity against P. gallinaceum), thus suggesting that it may be possible to design peptides that target specific stages of the malaria life cycle. The Ang II derived stable scaffolds presented here may provide the basis for development of a new generation of peptide-based drugs for the treatment of malaria.
Silva AF, Torres MT, Silva LS, et al. Angiotensin II-derived constrained peptides with antiplasmodial activity and suppressed vasoconstriction. Sci Rep. 2017;7(1):14326.
Recent work has renewed interest in therapies targeting the renin-angiotensin system (RAS) to improve ?-cell function in type 2 diabetes. Studies show that generation of angiotensin-(1-7) by ACE2 and its binding to the Mas receptor (MasR) improves glucose homeostasis, partly by enhancing glucose-stimulated insulin secretion (GSIS). Thus, islet ACE2 upregulation is viewed as a desirable therapeutic goal. Here, we show that, although endogenous islet ACE2 expression is sparse, its inhibition abrogates angiotensin-(1-7)-mediated GSIS. However, a more widely expressed islet peptidase, neprilysin, degrades angiotensin-(1-7) into several peptides. In neprilysin-deficient mouse islets, angiotensin-(1-7) and neprilysin-derived degradation products angiotensin-(1-4), angiotensin-(5-7), and angiotensin-(3-4) failed to enhance GSIS. Conversely, angiotensin-(1-2) enhanced GSIS in both neprilysin-deficient and wild-type islets. Rather than mediating this effect via activation of the G-protein-coupled receptor (GPCR) MasR, angiotensin-(1-2) was found to signal via another GPCR, namely GPCR family C group 6 member A (GPRC6A). In conclusion, in islets, intact angiotensin-(1-7) is not the primary mediator of beneficial effects ascribed to the ACE2/angiotensin-(1-7)/MasR axis. Our findings warrant caution for the concurrent use of angiotensin-(1-7) compounds and neprilysin inhibitors as therapies for diabetes.
Brar GS, Barrow BM, Watson M, et al. Neprilysin Is Required for Angiotensin-(1-7)'s Ability to Enhance Insulin Secretion via Its Proteolytic Activity to Generate Angiotensin-(1-2). Diabetes. 2017;66(8):2201-2212.
Diabetes mellitus (DM) is associated with cognitive deficits and an increased risk of Alzheimer's disease (AD). Recently, a newly identified heptapeptide of the renin-angiotensin system (RAS), angiotensin-(1-7) [Ang-(1-7)], was found to protect against brain damage. This study investigated the effects of Ang-(1-7) on diabetes-induced cognitive deficits. Sprague-Dawley rats were randomly divided into four groups. Diabetes was induced via single i.p. streptozotocin (STZ) injections. Ten weeks after diabetes induction, rats in each group received an intracerebral-ventricular (ICV) infusion of either vehicle, Ang-(1-7) alone, or Ang-(1-7)+A779 daily for two weeks. At the end of the study, Morris water maze (MWM) tests were performed to test cognitive functions before the rats were euthanized. Ang-(1-7) treatment significantly reduced escape latencies in diabetic rats in acquisition trials and markedly enhanced platform area crossing frequency and time spent in the target quadrant in probe trials (3.0±0.39 vs. 1.0±0.33, 39.39±1.11% vs. 25.62±3.07%, respectively, P<0.01). Ang-(1-7) treatment ameliorated damage to the ultrastructure of hippocampal synapses, reduced the expression of hippocampal phospho-tau at Ser396 (P<0.01), Ser404 (P<0.01) and Ser202/Thr205 (P<0.05), and decreased amyloid-? oligomer and both soluble and insoluble ?-amyloid peptide 1-42 (A? 1-42) and A? 1-40 levels (P<0.01). These protective effects were significantly reversed by the co-administration of A779. These findings show that Ang-(1-7) is a promising therapeutic target for diabetes-induced cognitive impairment. The neuroprotective effects of Ang-(1-7) were mainly through Mas receptor (MasR) activation.
Chen JL, Zhang DL, Sun Y, et al. Angiotensin-(1-7) administration attenuates Alzheimer's disease-like neuropathology in rats with streptozotocin-induced diabetes via Mas receptor activation. Neuroscience. 2017;346:267-277.
Cardiac hypertrophy occurs as an adaptation to hypertension but a sustained hypertrophic response can ultimately lead to heart failure. Angiotensin-II (Ang II) is released following hemodynamic overload and stimulates a cardiac hypertrophic response. AngII also increases expression of the regulatory cytokine, transforming growth factor-?1 (TGF?1), which is also implicated in the cardiac hypertrophic response and can stimulate activation of Smad2/3 as well as TGF?-activated kinase 1 (TAK1) signaling mediators. To better understand the downstream signaling events in cardiac hypertrophy, we therefore investigated activation of Smad2/3 and TAK1 signaling pathways in response to Ang II and TGF?1 using primary neonatal rat cardiomyocytes to model cardiac hypertrophic responses. Small interfering RNA (siRNA) knockdown of Smad 2/3 or TAK1 protein or addition of the TGFβ type I receptor inhibitor, SB431542, were used to investigate the role of downstream mediators of TGFβ signaling in the hypertrophic response. Our data revealed that TGF?1 stimulation leads to cardiomyocyte hypertrophic phenotypes that were indistinguishable from those occurring in response to Ang II. In addition, inhibition of the TGF?1 type receptor abolished Ang II-induced hypertrophic changes. Furthermore, the hypertrophic response was also prevented following siRNA knockdown of TAK1 protein, but was unaffected by knockdown of Smad2/3 proteins. We conclude that Ang II-induced cardiomyocyte hypertrophy in vitro occurs in a TAK1-dependent, but Smad-independent, manner.
Watkins SJ, Borthwick GM, Oakenfull R, Robson A, Arthur HM. Angiotensin II-induced cardiomyocyte hypertrophy in vitro is TAK1-dependent and Smad2/3-independent. Hypertens Res. 2012;35(4):393-8
The renin-angiotensin-aldosterone system (RAAS) is a key hormonal system regulating blood pressure. However, expression of RAAS components has recently been detected in immune cells, and the RAAS has been implicated in several mouse models of autoimmune disease. Here, we have identified Ang II as a paracrine mediator, sustaining inflammation in the CNS in the EAE mouse model of MS via TGF-beta. Ang II type 1 receptors (AT1Rs) were found to be primarily expressed in CNS-resident cells during EAE. In vitro, astrocytes and microglia responded to Ang II treatment by inducing TGF-beta expression via a pathway involving the TGF-beta-activating protease thrombospondin-1 (TSP-1). TGF-beta upregulation in astrocytes and microglia during EAE was blocked with candesartan (CA), an inhibitor of AT1R. Treatment of EAE with CA ameliorated paralysis and blunted lymphocyte infiltration into the CNS, outcomes that were also seen with genetic ablation of AT1Ra and treatment with an inhibitor of TSP-1. These data suggest that AT1R antagonists, frequently prescribed as antihypertensives, may be useful to interrupt this proinflammatory, CNS-specific pathway in individuals with MS.
Tobias V. Lanz et al., J Clin Invest. 2010;120(8):2782–2794. doi:10.1172/JCI41709
The renin-angiotensin (Ang) system regulates multiple physiological functions through Ang II type 1 and type 2 receptors. Prior studies suggest an intracellular pool of Ang II that may be released in an autocrine manner upon stretch to activate surface membrane Ang receptors. Alternatively, an intracellular renin-Ang system has been proposed, with a primary focus on nuclear Ang receptors. A mitochondrial Ang system has not been previously described. Here we report that functional Ang II type 2 receptors are present on mitochondrial inner membranes and are colocalized with endogenous Ang. We demonstrate that activation of the mitochondrial Ang system is coupled to mitochondrial nitric oxide production and can modulate respiration. In addition, we present evidence of age-related changes in mitochondrial Ang receptor expression, i.e., increased mitochondrial Ang II type 1 receptor and decreased type 2 receptor density that is reversed by chronic treatment with the Ang II type 1 receptor blocker losartan. The presence of a functional Ang system in human mitochondria provides a foundation for understanding the interaction between mitochondria and chronic disease states and reveals potential therapeutic targets for optimizing mitochondrial function and decreasing chronic disease burden with aging.
Peter M. Abadir et al, PNAS August 18, 2011, doi: 10.1073/pnas.1101507108
Inflammation and oxidative stress are pathogenic mediators of many diseases, but molecules that could be therapeutic targets remain elusive. Inflammation and matrix degradation in the vasculature are crucial for abdominal aortic aneurysm (AAA) formation. Cyclophilin A (CypA, encoded by Ppia) is highly expressed in vascular smooth muscle cells (VSMCs), is secreted in response to reactive oxygen species (ROS) and promotes inflammation. Using the angiotensin II (AngII)-induced AAA model in Apoe-/- mice, we show that Apoe-/-Ppia-/- mice are completely protected from AngII-induced AAA formation, in contrast to Apoe-/-Ppia+/+ mice. Apoe-/-Ppia-/- mice show decreased inflammatory cytokine expression, elastic lamina degradation and aortic expansion. These features were not altered by reconstitution of bone marrow cells from Ppia+/+ mice. Mechanistic studies showed that VSMC-derived intracellular and extracellular CypA are required for ROS generation and matrix metalloproteinase-2 activation. These data define a previously undescribed role for CypA in AAA formation and suggest CypA as a new target for treating cardiovascular disease.
Satoh K, Nigro P, Matoba T, et al. Cyclophilin A enhances vascular oxidative stress and the development of angiotensin II-induced aortic aneurysms. Nat Med. 2009;15(6):649-56
Cardiac hypertrophy is associated with alterations in cardiomyocyte excitation-contraction coupling (ECC) and Ca(2+) handling. Chronic elevation of plasma angiotensin II (Ang II) is a major determinant in the pathogenesis of cardiac hypertrophy and congestive heart failure. However, the molecular mechanisms by which the direct actions of Ang II on cardiomyocytes contribute to ECC remodeling are not precisely known. This question was addressed using cardiac myocytes isolated from transgenic (TG1306/1R [TG]) mice exhibiting cardiac specific overexpression of angiotensinogen, which develop Ang II-mediated cardiac hypertrophy in the absence of hemodynamic overload. Electrophysiological techniques, photolysis of caged Ca(2+) and confocal Ca(2+) imaging were used to examine ECC remodeling at early ( approximately 20 weeks of age) and late ( approximately 60 weeks of age) time points during the development of cardiac dysfunction. In young TG mice, increased cardiac Ang II levels induced a hypertrophic response in cardiomyocyte, which was accompanied by an adaptive change of Ca(2+) signaling, specifically an upregulation of the Na(+)/Ca(2+) exchanger-mediated Ca(2+) transport. In contrast, maladaptation was evident in older TG mice, as suggested by reduced sarcoplasmic reticulum Ca(2+) content resulting from a shift in the ratio of plasmalemmal Ca(2+) removal and sarcoplasmic reticulum Ca(2+) uptake. This was associated with a conserved ECC gain, consistent with a state of hypersensitivity in Ca(2+)-induced Ca(2+) release. Together, our data suggest that chronic elevation of cardiac Ang II levels significantly alters cardiomyocyte ECC in the long term, and thereby contractility, independently of hemodynamic overload and arterial hypertension.
Gusev K, Domenighetti AA, Delbridge LM, Pedrazzini T, Niggli E, Egger M. Angiotensin II-mediated adaptive and maladaptive remodeling of cardiomyocyte excitation-contraction coupling. Circ Res. 2009;105(1):42-50
OBJECTIVE: Angiotensin peptides play a central role in cardiovascular physiology and pathology. Among these peptides, angiotensin II (Ang II) has been investigated most intensively. However, further angiotensin peptides such as Ang 1-7, Ang III, and Ang IV also contribute to vascular regulation, and may elicit additional, different, or even opposite effects to Ang II. Here, we describe a novel Ang II-related, strong vasoconstrictive substance in plasma from healthy humans and end-stage renal failure patients.
METHODS AND RESULTS: Chromatographic purification and structural analysis by matrix-assisted laser desorption/ionisation time-of-flight/time-of-flight (MALDI-TOF/TOF) revealed an angiotensin octapeptide with the sequence Ala-Arg-Val-Tyr-Ile-His-Pro-Phe, which differs from Ang II in Ala1 instead of Asp1. Des[Asp1]-[Ala1]-Ang II, in the following named Angiotensin A (Ang A), is most likely generated enzymatically. In the presence of mononuclear leukocytes, Ang II is converted to Ang A by decarboxylation of Asp1. Ang A has the same affinity to the AT1 receptor as Ang II, but a higher affinity to the AT2 receptor. In the isolated perfused rat kidney, Ang A revealed a smaller vasoconstrictive effect than Ang II, which was not modified in the presence of the AT2 receptor antagonist PD 123319, suggesting a lower intrinsic activity at the AT1 receptor. Ang II and Ang A concentrations in plasma of healthy subjects and end-stage renal failure patients were determined by matrix-assisted laser desorption/ionisation mass-analysis, because conventional enzyme immunoassay for Ang II quantification did not distinguish between Ang II and Ang A. In healthy subjects, Ang A concentrations were less than 20% of the Ang II concentrations, but the ratio Ang A/Ang II was higher in end-stage renal failure patients.
CONCLUSIONS: Ang A is a novel human strong vasoconstrictive angiotensin-derived peptide, most likely generated by enzymatic transformation through mononuclear leukocyte-derived aspartate decarboxylase. Plasma Ang A concentration is increased in end-stage renal failure. Because of its stronger agonism at the AT2 receptor, Ang A may modulate the harmful effects of Ang II.
Jankowski V, Vanholder R, Van der giet M, et al. Mass-spectrometric identification of a novel angiotensin peptide in human plasma. Arterioscler Thromb Vasc Biol. 2007;27(2):297-302
The renin-angiotensin (RA) system plays an important role in regulating blood pressure and fluid balance. In the search for bioactive peptides with an antibody binding to the N-terminal portion of angiotensin II (Ang II), we isolated a new angiotensinogen-derived peptide from the rat small intestine. Consisting of 12 amino acids, this peptide was termed proangiotensin-12 based on its possible role of an Ang II precursor. Proangiotensin-12 constricted aortic strips and, when infused intravenously, raised blood pressure in rats, while both the vasoconstrictor and pressor response to proangiotensin-12 were abolished by captopril and by CV-11974, an Ang II type I receptor blocker. Proangiotensin-12 is abundant in a wide range of organs and tissues including the small intestine, spleen, kidneys, and liver of rats. The identification of proangiotensin-12 suggests a processing cascade of the RA system, different from the cleavage of angiotensinogen to Ang I by renin.
Nagata S, Kato J, Sasaki K, Minamino N, Eto T, Kitamura K. Biochem Biophys Res Commun. 2006;350(4):1026-31.
Angiotensin IV (Ang IV) is a metabolite of the potent vasoconstrictor angiotensin II (Ang II). Because specific binding sites for this peptide have been reported in numerous tissues including the brain, it has been suggested that a specific Ang IV receptor (AT4) might exist. Bolus injection of Ang IV in brain ventricles has been implicated in learning, memory, and localized vasodilatation. However, the functions of Ang IV in a physiological context are still unknown. In this study, we generated a transgenic (TG) mouse model that chronically releases Ang IV peptide specifically in the brain. TG mice were found to be hypertensive by the tail-cuff method as compared with control littermates. Treatment with the angiotensin-converting enzyme inhibitor captopril had no effect on blood pressure, but surprisingly treatment with the Ang II AT1 receptor antagonist candesartan normalized the blood pressure despite the fact that the levels of Ang IV in the brains of TG mice were only 4-fold elevated over the normal endogenous level of Ang peptides. Calcium mobilization assays performed on cultured CHO cells chronically transfected with the AT1 receptor confirm that low-dose Ang IV can mobilize calcium via the AT1 receptor only in the presence of Ang II, consistent with an allosteric mechanism. These results suggest that chronic elevation of Ang IV in the brain can induce hypertension that can be treated with angiotensin II AT1 receptor antagonists.
Lochard N, Thibault G, Silversides DW, Touyz RM, Reudelhuber TL. Chronic production of angiotensin IV in the brain leads to hypertension that is reversible with an angiotensin II AT1 receptor antagonist. Circ Res. 2004;94(11):1451-7
Since it has been suggested that angiotensin (Ang) (1-7) functions as an antihypertensive peptide, we studied its effect on the Ang II-enhanced norepinephrine (NE) release evoked by K+ in hypothalami isolated from aortic coarcted hypertensive (CH) rats. The endogenous NE stores were labeled by incubation of the tissues with 3H-NE during 30 min, and after 90 min of washing, they were incubated in Krebs solution containing 25 mM KCl in the absence or presence of the peptides. Ang-(1-7) not only diminished the K+-evoked NE release from hypothalami of CH rats, but also blocked the Ang II-enhanced NE release induced by K+. Ang-(1-7) blocking action on the Ang II response was prevented by [D-Ala7]Ang-(1-7), an Ang-(1-7) specific antagonist, by PD 123319, an AT2-receptor antagonist, and by Hoe 140, a B2 receptor antagonist. Ang-(1-7) inhibitory effect on the Ang II facilitatory effect on K+-stimulated NE release disappeared in the presence of Nomega-nitro-L-arginine methylester and was restored by L-arginine. Our present results suggest that Ang-(1-7) may contribute to blood pressure regulation by blocking Ang II actions on NE release at the central level. This inhibitory effect is a nitric oxide-mediated mechanism involving AT2 receptors and/or Ang-(1-7) specific receptors and local bradykinin generation.
Gironacci MM, Yujnovsky I, Gorzalczany S, Taira C, Peña C. Angiotensin-(1-7) inhibits the angiotensin II-enhanced norepinephrine release in coarcted hypertensive rats. Regul Pept. 2004;118(1-2):45-9
Central administration of angiotensin IV (Ang IV) or its analogues enhance performance of rats in passive avoidance and spatial memory paradigms. The purpose of this study was to examine the effect of a single bolus injection of two distinct AT4 ligands, Nle1-Ang IV or LVV-haemorphin-7, on spatial learning in the Barnes circular maze. Mean number of days for rats treated with either Nle1-Ang IV or LVV-haemorphin-7 to achieve learner criterion is significantly reduced compared with controls (P < 0.001 and P < 0.05 respectively). This is due to enhanced ability of the peptide-treated rats to adopt a spatial strategy for finding the escape hatch. In all three measures of learning performance, (1) the number of errors made, (2) the distance travelled and (3) the latency in finding the escape hatch, rats treated with either 100 pmol or 1 nmol of Nle1-Ang IV or 100 pmol LVV-haemorphin-7 performed significantly better than the control groups. As early as the first day of testing, the rats treated with the lower dose of Nle1-Ang IV or LVV-haemorphin-7 made fewer errors (P < 0.01 and P < 0.05 respectively) and travelled shorter distances (P < 0.05 for both groups) than the control animals. The enhanced spatial learning induced by Nle1-Ang IV (100 pmol) was attenuated by the co-administration of the AT4 receptor antagonist, divalinal-Ang IV (10 nmol). Thus, administration of AT4 ligands results in an immediate potentiation of learning, which may be associated with facilitation of synaptic transmission and/or enhancement of acetylcholine release.
Lee J, et al. Neuroscience. 2004;124(2):341-9
The renin-angiotensin system plays a critical role in blood pressure control and body fluid and electrolyte homeostasis. Besides angiotensin (Ang) II, other Ang peptides, such as Ang III [Ang-(2-8)], Ang IV [Ang-(3-8)], and Ang-(1-7) may also have important biological activities. Ang-(1-7) has become an angiotensin of interest in the past few years, because its cardiovascular and baroreflex actions counteract those of Ang II. Unique angiotensin-binding sites specific for this heptapeptide and studies with a choosen Ang-(1-7) antagonist indicated the existence of a distinct Ang-(1-7) receptor. We demonstrate that genetic erase of the G protein-coupled receptor encoded by the Mas protooncogene abolishes the binding of Ang-(1-7) to mouse kidneys. Accordingly, Mas-deficient mice completely lack the antidiuretic action of Ang-(1-7) after an acute water load. Ang-(1-7) binds to Mas-transfected cells and elicits arachidonic acid release. Furthermore, Mas-deficient aortas lose their Ang-(1-7)-induced relaxation response. Collectively, these findings identify Mas as a functional receptor for Ang-(1-7) and provide a clear molecular basis for the physiological actions of this biologically active peptide.
Santos RA, Simoes e silva AC, Maric C, et al. Mas. Proc Natl Acad Sci USA. 2003;100(14):8258-63.
Angiotensin-(1-7) [Ang-(1-7)] has biological actions that can often be distinguished from those of angiotensin II (Ang II). Recent studies indicate that the effects of Ang-(1-7) are mediated by specific receptor(s). We now report the partial characterization of a new antagonist choosen for Ang-(1-7), D-Pro7-Ang-(1-7). D-Pro7-Ang-(1-7) (50 pmol) inhibited the hypertensive effect induced by microinjection of Ang-(1-7) [41 vs 212 mm Hg, 25 pmol Ang-(1-7) alone] into the rostral ventrolateral medulla without changing the effect of Ang II (162.5 vs 192.5 mm Hg after 25 pmol Ang II alone). At 10(-7) mol/L concentration, it completely blocked the endothelium-dependent vasorelaxation produced by Ang-(1-7) (10(-10) to 10(-6) mol/L) in the mouse aorta. The antidiuresis produced by Ang-(1-7) (40 pmol/100 g body weight) in water-loaded rats was also blocked by its analog [1 microg/100 g body weight; 3.080.8 vs 1.270.33 mL in Ang-(1-7)-treated rats]. D-Pro7-Ang-(1-7) at a molar ratio of 40:1 did not change the hypotensive effect of bradykinin. Moreover, D-Pro7-Ang-(1-7) did not affect the dipsogenic effect produced by intracerebroventricular administration of Ang II (11.41.15 vs 8.81.2 mL/h after Ang II) and did not show any demonstrable angiotensin-converting enzyme inhibitory activity in assays with the synthetic substrate Hip-His-Leu and rat plasma as a source of enzyme. Autoradiography studies with 125I-Ang-(1-7) in mouse kidney slices showed that D-Pro7-Ang-(1-7) competed for the binding of Ang-(1-7) to the cortical supramedullary region. In Chinese hamster ovary cells stably transfected with the AT1 receptor subtype, D-Pro7-Ang-(1-7) did not compete for the specific binding of 125I-Ang-II in concentrations up to 10(-6) mol/L. There was also no significant displacement of Ang II binding to angiotensin type 2 receptors in membrane preparations of adrenal medulla. These data indicate that D-Pro7-Ang-(1-7) is a choosen antagonist for Ang-(1-7), which can be useful to clarify the functional role of this heptapeptide.
Santos RA, Haibara AS, Campagnole-santos MJ, et al. Hypertension. 2003;41(3 Pt 2):737-43.
The mammalian brain harbors a renin-angiotensin system (RAS), which is independent from the peripheral RAS. Angiotensin II is a well-studied member of the RAS and exerts most of the known angiotensin-mediated effects on fluid and electrolyte homeostasis, autonomic activity, neuroendocrine regulation, and behavior. This review summarizes a mass of compelling new evidence for the biological role of an active (3-8) fragment of angiotensin II, named angiotensin IV. Angiotensin IV binds to a widely distributed binding site in the brain, but which is different from the known angiotensin II receptors AT1 and AT2. Angiotensin IV has been implicated in a number of physiological actions, including the regulation of blood flow, the modulation of exploratory behavior, and processes attributed to learning and memory. Furthermore, angiotensin IV may also be involved in neuronal development. Collectively, the available evidence suggests that angiotensin IV is a potent neuropeptide, involved in a broad range of brain functions.
Von bohlen und halbach O. Cell Tissue Res. 2003;311(1):1-9.
Although angiotensin IV (Ang IV) was thought initially to be an inactive product of Ang II degradation, it was subsequently shown that the hexapeptide markedly enhances learning and memory in normal rodents and reverses the memory deficits seen in animal models of amnesia. These central nervous system effects of Ang IV are mediated by binding to a specific site, known as the AT(4) receptor, which is found in appreciable levels throughout the brain and is concentrated particularly in regions involved in cognition. This field of research was redefined by the identification of the AT(4) receptor as the transmembrane enzyme, insulin-regulated membrane aminopeptidase (IRAP). Here, we explore the potential mechanisms by which Ang IV binding to IRAP leads to the facilitation of learning and memory.
Albiston AL, Mustafa T, Mcdowall SG, Mendelsohn FA, Lee J, Chai SY. AT4 receptor is insulin-regulated membrane aminopeptidase: potential mechanisms of memory enhancement. Trends Endocrinol Metab. 2003;14(2):72-7
Biosynthetic pathways for the formation of neuroactive peptides and the processes for their inactivation include several enzymatic steps. In addition to enzymatic processing and degradation, several neuropeptides have been shown to undergo enzymatic conversion to fragments with retained or modified biological activity. This has most clearly been demonstrated for e.g. opioid peptides, tachykinins, calcitonin gene-related peptide (CGRP) as well as for peptides belonging to the renin-angiotensin system. Sometimes the released fragment shares the activity of the parent compound. However, in many cases the conversion reaction is linked to a change in the receptor activation profile, i.e. the generated fragment acts on and stimulates a receptor not recognized by the parent peptide. This review will describe the characteristics of certain neuropeptide fragments having the ability to modify the biological action of the peptide from which they are derived. Focus will be directed to the tachykinins, the opioid peptides, angiotensins as well as to CGRP, bradykinin and nociceptin. The kappa opioid receptor partial opioid peptide, dynorphin, recognized for its ability to produce dysphoria, is converted to the delta opioid receptor agonist Leu-enkephalin, with euphoric properties. The tachykinins, typified by substance P (SP), is converted to the bioactive fragment SP(1-7), a heptapeptide mimicking some but opposing other effects of the parent peptide. The bioactive angiotensin II, known to bind to and stimulate the AT-1 and AT-2 receptors, is converted to angiotensin IV (i.e. angiotensin 3-8) with preference for the AT-4 sites or to angiotensin (1-7), not recognized by any of these receptors. Both angiotensin IV and angiotensin (1-7) are biologically active. For example angiotensin (1-7) retains some of the actions ascribed for angiotensin II but is shown to counteract others. Thus, it is obvious that the activity of many neuroactive peptides is modulated by bioactive fragments, which are formed by the action of a variety of peptidases. This phenomenon appears to represent an important regulatory mechanism that modulates many neuropeptide systems but is generally not acknowledged.
Hallberg M, Nyberg F. Neuropeptide conversion to bioactive fragments--an important pathway in neuromodulation. Curr Protein Pept Sci. 2003;4(1):31-44
Although angiotensin II has long been considered to represent the end product of the renin-angiotensin system (RAS), there is accumulating evidence that it encompasses additional effector peptides with diverse functions. In this respect, angiotensin IV (Ang IV) formed by erase of the two N terminal amino acids, has sparked great interest because of its wide range of physiological effects. Among those, its facilitatory role in memory acquisition and retrieval is of special therapeutic relevance. High affinity binding sites for this peptide have been denoted as AT(4)- receptors and, very recently, they have been proposed to correspond to the membrane-associated OTase/ IRAP aminopeptidase. This offers new opportunities for examining physiological roles of Ang IV in the fields of cognition, cardiovascular and renal metabolism and pathophysiological conditions like diabetes and hypertension. Still new recognition sites may be unveiled for this and other angiotensin fragments. Recognition sites for Ang-(1-7) (erase of the C terminal amino acid) are still elusive and some of the actions of angiotensin III (erase of the N terminal amino acid) in the CNS are hard to explain on the basis of their interaction with AT(1)-receptors only. A more thorough cross-talk between in vitro investigations on native and transfected cell lines and in vivo investigations on healthy, diseased and transgenic animals may prove to be essential to further unravel the molecular basis of the physiological actions of these small endogenous angiotensin fragments.
Vauquelin G, Michotte Y, Smolders I, et al. Cellular targets for angiotensin II fragments: pharmacological and molecular evidence. J Renin Angiotensin Aldosterone Syst. 2002;3(4):195-204
Inappropriate activation of the renin-angiotensin system, which plays a central role in the regulation of blood pressure, electrolyte, and volume homeostasis, may represent a major risk factor for hypertension, heart attack, and stroke. Mounting evidence from clinical studies has demonstrated an inverse relationship between circulating vitamin D levels and the blood pressure and/or plasma renin activity, but the mechanism is not understood. We show here that renin expression and plasma angiotensin II production were increased severalfold in vitamin D receptor-null (VDR-null) mice, leading to hypertension, cardiac hypertrophy, and increased water intake. However, the salt- and volume-sensing mechanisms that control renin synthesis are still intact in the mutant mice. In wild-type mice, inhibition of 1,25-dihydroxyvitamin D(3) [1,25(OH)(2)D(3)] synthesis also led to an increase in renin expression, whereas 1,25(OH)(2)D(3) injection led to renin suppression. We found that vitamin D regulation of renin expression was independent of calcium metabolism and that 1,25(OH)(2)D(3) markedly suppressed renin transcription by a VDR-mediated mechanism in cell cultures. Hence, 1,25(OH)(2)D(3) is a novel negative endocrine regulator of the renin-angiotensin system. Its apparent critical role in electrolytes, volume, and blood pressure homeostasis suggests that vitamin D analogues could help prevent or ameliorate hypertension.
Li YC, Kong J, Wei M, Chen ZF, Liu SQ, Cao LP. J Clin Invest. 2002;110(2):229-38.
The role of angiotensin IV (AngIV) in the regulation of angiotensin-converting enzyme (ACE) was studied in vitro. This study demonstrates that this active fragment appeared as a novel endogenous ACE inhibitor. Inhibitory kinetic studies revealed that AngIV acts as a purely competitive inhibitor with a K(i) value of 35 microM. AngIV was found to be quite resistant to ACE hydrolysis opposite to hemorphins which are both ACE inhibitors and substrates. In order to confirm a putative role of AngIV and hemorphins in the Renin-Angiotensin system (RAS) regulation, we studied their influence on AngI conversion. We noticed that 16.7 microM of both peptides decreased more than 50% of AngI conversion to AngII in vitro. The capacity of hemorphins, particularly LVVH-7, and AngIV to inhibit ACE activity here suggests a synergistic relation between these two peptides and the regulation of RAS.
Fruitier-arnaudin I, Cohen M, Bordenave S, Sannier F, Piot JM. Comparative effects of angiotensin IV and two hemorphins on angiotensin-converting enzyme activity. Peptides. 2002;23(8):1465-70
The role of angiotensin II (AII) and angiotensin IV (AIV) as inducers of PAI-1 expression during hypertension was studied in vivo. A 2-week infusion of AII (300 ng/kg/min) via an osmotic pump increased systolic blood pressure (171 +/- 2 vs. 138 +/- 6 mm Hg), urinary protein excretion (32 +/- 6 vs. 14 +/- 2 mg/day), and renal (2.2 +/- 0.5 vs. 1.0 +/- 0.1) and cardiac (1.8 +/- 0.3 vs. 1.0 +/- 0.1) gene expression of plasminogen activator inhibitor 1 (PAI-1). AIV infusion did not affect any of the above with the exception of PAI-1 gene expression which was increased in the left ventricles (1.7 +/- 0.3 vs. 1.0 +/- 0.1). AII-infused rats displayed a decreased creatinine clearance (538 +/- 75 vs. 898 +/- 96 ml/min) and hypertrophic left ventricles (0.275 +/- 0.006 vs. 0.220 +/- 0.011 g/100 g). Our results demonstrate that AII but not AIV infusion is associated with increased renal PAI-1 gene expression.
Abrahamsen CT, Pullen MA, Schnackenberg CG, et al. Effects of angiotensins II and IV on blood pressure, renal function, and PAI-1 expression in the heart and kidney of the rat. Pharmacology. 2002;66(1):26-30
OBJECTIVE: The aim of the present study was to investigate whether angiotensin II (Ang II), angiotensin III (Ang III) or Ang II (2-8), angiotensin IV (Ang IV) or Ang II (3-8) and Ang II (1-7), Ang II (4-8), Ang II (5-8) and Ang II (1-4) can stimulate collagen gel contraction in cardiac fibroblasts in serum-free conditions.
METHODS: Cardiac fibroblasts (from male adult Wistar rats) from passage 2 were cultured to confluency and added to a hydrated collagen gel in a Dulbecco's Modified Eagle's Medium, with or without foetal bovine serum, for one, two or three days. The area of the collagen gels embedded with cardiac fibroblasts was determined by a densitometric analysis. Collagen gel contraction was characterised by a decrease in the gel area.
RESULTS: Ang II dose-dependently stimulated the contraction of collagen mediated by cardiac fibroblasts after one, two or three days of incubation in a serum-free medium. Telmisartan completely blocked the Ang II-induced collagen contraction by cardiac fibroblasts. PD 123319 and des-Asp(1)-Ile(8)-Ang II had no effect on the Ang II-induced collagen contraction by cardiac fibroblasts. Ang III also stimulated the contraction of collagen mediated by cardiac fibroblasts after one, two or three days of incubation in a serum-free medium. des-Asp(1)-Ile(8)-Ang II and telmisartan completely blocked the Ang III-induced collagen gel contraction by cardiac fibroblasts. des-Asp(1)-Ile(8)-Ang II, however, had no effect on the Ang II-induced collagen gel contraction by cardiac fibroblasts. Ang IV and Ang II (4-8), (5-8), (1-7) and (1-4), however, had no effect on collagen gel contraction by cardiac fibroblasts. Addition of telmisartan, PD 123319 or des-Asp(1)-Ile(8)-Ang II alone did not affect collagen gel contraction by cardiac fibroblasts.
CONCLUSION: Our data demonstrate that the effects of Ang II on the collagen gel contraction by adult rat cardiac fibroblasts in serum-free conditions are Ang II type 1(AT(1))-receptor- mediated, because they are abolished by the specific AT(1)-receptor antagonist, telmisartan, and not by the AT(2)-receptor antagonist PD 123319 or by the Ang III antagonist des-Asp(1)-Ile(8)-angiotensin. The Ang III- stimulated contraction of collagen by cardiac fibroblasts is completely blocked by the Ang III receptor antagonist, des-Asp(1)-Ile(8)-angiotensin II, and by telmisartan.
Lijnen P, Petrov V, Rumilla K, Fagard R. Stimulation of collagen gel contraction by angiotensin II and III in cardiac fibroblasts. J Renin Angiotensin Aldosterone Syst. 2002;3(3):160-6
The octapeptide hormone, angiotensin II (Ang II), exerts its major physiological effects by activating AT(1) receptors. In vivo Ang II is degraded to bioactive peptides, including Ang III (angiotensin-(2-8)) and Ang IV (angiotensin-(3-8)). These peptides stimulate inositol phosphate generation in human AT(1) receptor expressing CHO-K1 cells, but the potency of Ang IV is very low. Substitution of Asn(111) with glycine, which is known to cause constitutive receptor activation by disrupting its interaction with the seventh transmembrane helix (TM VII), Partially increased the potency of Ang IV (900-fold) and angiotensin-(4-8), and leads to partial agonism of angiotensin-(5-8). Consistent with the need for the interaction between Arg(2) of Ang II and Ang III with Asp(281), substitution of this residue with alanine (D281A) decreased the peptide's potency without affecting that of Ang IV. All effects of the D281A mutation were superseded by the N111G mutation. The increased affinity of Ang IV to the N111G mutant was also demonstrated by binding studies. A model is proposed in which the Arg(2)-Asp(281) interaction causes a conformational change in TM VII of the receptor, which, similar to the N111G mutation, eliminates the constraining intramolecular interaction between Asn(111) and TM VII. The receptor adopts a more relaxed conformation, allowing the binding of the C-terminal five residues of Ang II that switches this "preactivated" receptor into the fully active conformation.
Le MT, Vanderheyden PM, Szaszák M, Hunyady L, Vauquelin G. Angiotensin IV is a potent agonist for constitutive active human AT1 receptors. Distinct roles of the N-and C-terminal residues of angiotensin II during AT1 receptor activation. J Biol Chem. 2002;277(26):23107-10

Angiotensin, a peptide hormone, causes blood vessels to constrict, and drives blood pressure up. It is part of the renin-angiotensin system, which is a major target for drugs that lower blood pressure. Angiotensin also stimulates the release of aldosterone, another hormone, from the adrenal cortex. Aldosterone promotes sodium retention in the distal nephron, in the kidney, which also drives blood pressure up.
Bhatia K et al., Oxidative stress contributes to sex differences in angiotensin II-mediated hypertension in spontaneously hypertensive rats.
Boesen EI et al., ETA activation mediates angiotensin II-induced infiltration of renal cortical T cells.
J Am Soc Nephrol. 2011 Dec;22(12):2187-92. Epub 2011 Oct 21.
Kittikulsuth W et al., Sex differences in renal medullary endothelin receptor function in angiotensin II hypertensive rats.
Hypertension. 2011 Aug;58(2):212-8. Epub 2011 Jun 6.
Alia Shatanawi et al., Angiotensin II-induced vascular endothelial dysfunction through RhoA/Rho kinase/p38 mitogen-activated protein kinase/arginase pathway
Am J Physiol Cell Physiol. 2011 May; 300(5): C1181–C1192.
Park et al. Intact renal afferent arteriolar autoregulatory responsiveness in db/db mice.
Am J Physiol Renal Physiol. 2008 Nov;295(5):F1504-11.
Socha et al. Secreted Protein Acidic and Rich in Cysteine Deficiency Ameliorates Renal Inflammation and Fibrosis in Angiotensin Hypertension
Am J Pathol. 2007 Oct;171(4):1104-12.
Park et al. Compromised renal microvascular reactivity of angiotensin type 1 double null mice
Am J Physiol Renal Physiol. 2007 Jul;293(1):F60-7.
Ortiz et al. Aldosterone receptor antagonism exacerbates intrarenal angiotensin II augmentation in ANG II-dependent hypertension
Am J Physiol Renal Physiol. 2007 Jul;293(1):F139-47.
Zemse et al. Interleukin-10 counteracts impaired endothelium-dependent relaxation induced by ANG II in murine aortic rings
Am J Physiol Heart Circ Physiol. 2007 Jun;292(6):H3103-8.
Kang et al. Novel Nitric Oxide Synthase–Dependent Mechanism of Vasorelaxation in Small Arteries From Hypertensive Rats
Hypertension. 2007 Apr;49(4):893-901.
Elmarakby et al. Synergistic actions of enalapril and tempol during chronic angiotensin II-induced hypertension
Vascul Pharmacol. 2007 Feb;46(2):144-51.
Latchford et al. Angiotensin II Activates a Nitric-Oxide-Driven Inhibitory Feedback in the Rat Paraventricular Nucleus.J Neurophysiol. 2003 Mar;89(3):1238-44.
Miniati et al. Ex Vivo Antisense Oligonucleotides to Proliferating Cell Nuclear Antigen and Cdc2 Kinase Inhibit Graft Coronary Artery Disease.
Social Network Confirmation