
The anti-inflammatory properties of annexin-1 peptides have been largely ascribed to their powerful antineutrophil actions in vivo. We have recently reported that the N-terminal fragment of annexin-1, Anx-12–26, preserves contractile function of cardiac muscle in vitro. The aim of the present study was to determine if Anx-12–26 elicits protective actions specifically on the cardiac myocyte (in the absence of neutrophils), using a model of metabolic inhibition to simulate ischaemia. Metabolic inhibition of cardiac myocytes (4 h incubation at 37°C in HEPES-containing buffer supplemented with 2-deoxy-d-glucose, d,l-lactic acid and pH adjusted to 6.5) followed by 2.5 h recovery in normal medium markedly increased creatine kinase (CK) and lactate dehydrogenase (LDH) levels by 179±39 and 26±7 IU/L (both n=40, P<0.001), respectively. However, cellular injury was significantly decreased when Anx-12–26 (0.3 ?M) was present during metabolic inhibition, CK by 74±10% and LDH by 71±6% (both n=31, P<0.001), respectively. Boc 2 (10 ?M), a nonselective formyl peptide receptor antagonist, present during metabolic inhibition, abolished the cardioprotective effect of Anx-12–26. Addition of chelerythrine (10 ?M), 5-hydroxydecanoate (500 ?M) or SB202190 (1 ?M) during metabolic inhibition also abolished Anx-12–26-induced cardioprotection. Cellular injury induced by metabolic inhibition was also largely prevented when myocytes were incubated with Anx-12–26 for 5 min with 10 min recovery prior to the insult, or when Anx-12–26 was present only during the recovery period following drug-free metabolic inhibition. In conclusion, the annexin-1 peptide Anx-12–26 potently prevents cardiac myocyte injury induced by metabolic inhibition, an action that was dependent at least in part on the activation of the formyl peptide receptor family of G-protein-coupled receptors, protein kinase C, p38 mitogen-activated protein kinase and ATP-sensitive potassium channels.
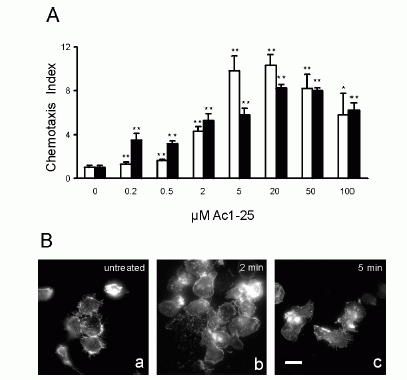
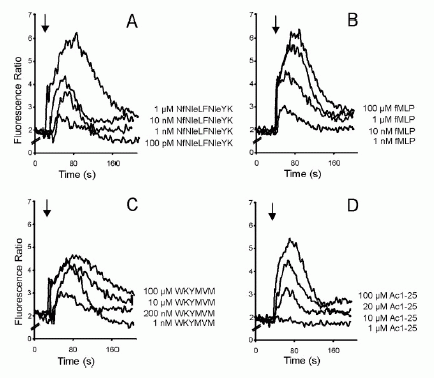
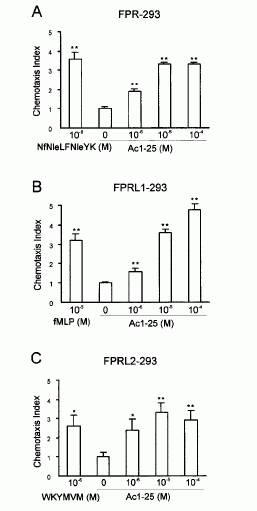
FIGURE 1. Directional migration of human peripheral blood granulocytes and monocytes. A, Chemotactic activity of the annexin 1 peptide Ac1-25 for human granulocytes and monocytes. Migration of granulocytes () or monocytes (f) in response to different peptide concentrations is presented as the average chemotaxis index SEM of triplicate wells. , p0.05, , p 0.01. B, The annexin 1 peptide Ac1-25 alters neutrophil morphology and F-actin distribution. Rhodamine-phalloidin staining of F-actin in unstimulated granulocytes (a) or cells treated with 50 M Ac1-25 for 2 min (b) or 5 min (c). Cells were fixed following treatment and analyzed by fluorescence microscopy. Note that Ac1-25 causes remodeling of the actin cytoskeleton, cell polarization, and spreading. Bar represents 10 m. .01. Ernst S, et al. An annexin 1 N-terminal Peptide activates leukocytes by triggering different members of the formyl Peptide receptor family. FIGURE 2. Dose-response curves of intracellular Ca2 mobilization in monocytes. Monocytes loaded with fura 2-AM were stimulated with increasing concentrations of the fMLP analog NfNleLFNleYK (A), fMLP (B), WKYMVM (C) or Ac1-25 (D). Fluorescence ratios of representative recordings are presented. Arrows indicate agonist addition. .01. Ernst S, et al. An annexin 1 N-terminal Peptide activates leukocytes by triggering different members of the formyl Peptide receptor family. FIGURE 3. Annexin 1 can use all members of the FPR family to induce chemotaxis. Various concentrations of chemoattractant were added in the lower wells of a chemotaxis chamber. HEK 293 cells stably expressing FPR, FPRL1, or FPRL2, respectively (106 cells/ml), were placed in the upper wells. Following incubation the number of cells that had migrated toward the chemoattractant source was determined as described in Materials and Methods. Cell migration is expressed as the average chemotaxis index (mean SEM) of six wells. A, Migration of FPR-293 cells in response to Ac1-25 or the f MLP analog NfNleLFNleYK is shown. B, The migration of FPRL1-293 cells in response to Ac1-25 or f MLP and (C) migration of FPRL2-293 cells in response to Ac1-25 or the synthetic peptide WKYMVM. , p 0.05, , p 0 .01. Ernst S, et al. An annexin 1 N-terminal Peptide activates leukocytes by triggering different members of the formyl Peptide receptor family.
Ritchie RH, Gordon JM, Woodman OL, Cao AH, Dusting GJ. Br J Pharmacol. 2005;145(4):495-502.
The human N-formyl peptide receptor (FPR) is a key modulator of chemotaxis directing granulocytes toward sites of bacterial infections. FPR is the founding member of a subfamily of G protein-coupled receptors thought to function in inflammatory processes. The other two members, FPR-like (FPRL)1 and FPRL2, have a greatly reduced affinity for bacterial peptides or do not bind them at all, with FPRL2 being considered an orphan receptor so far. In this study we show that a peptide derived from the N-terminal domain of the anti-inflammatory protein annexin 1 (lipocortin 1) can activate all three FPR family members at similar concentrations. The annexin 1 peptide initiates chemotactic responses in human monocytes that express all three FPR family members and also desensitizes the cells toward subsequent stimulation with bacterial peptide agonists. Experiments using HEK 293 cells stably expressing a single FPR family member reveal that all three receptors can be activated and desensitized by the N-terminal annexin 1 peptide. These observations identify the annexin 1 peptide as the first endogenous ligand of FPRL2 and indicate that annexin 1 participates in regulating leukocyte emigration into inflamed tissue by activating and desensitizing different receptors of the FPR family.
Ernst S, Lange C, Wilbers A, Goebeler V, Gerke V, Rescher U. An annexin 1 N-terminal peptide activates leukocytes by triggering different members of the formyl peptide receptor family. J Immunol. 2004;172(12):7669-76.
BACKGROUND: Annexin-1 (ANXA1, lipocortin 1) is a pleiotrophic protein produced by many cell types including peripheral blood leucocytes. Although it has been shown to inhibit "macroscopic" inflammatory processes in animal models, its direct effects on antigen-activated human T cells have not been studied. OBJECTIVE: To test the hypothesis that ANXA1-derived peptides inhibit antigen-driven prototype Th1 and Th2-type human T cell responses of clinical relevance and lectin-driven responses in vitro.
METHODS: Peripheral blood mononuclear cells (PBMC) were isolated from 14 atopic subjects sensitized to house dust mite allergen (Dermatophagoides pteronyssinus, Der p) and purified protein derivative (PPD) of Mycobacterium tuberculosis. PBMC (1 x 106/mL) were cultured with phytohaemagglutinin (PHA; 5 microg/mL; 4 days), Der p (25 microg/mL; 6 days), PPD (10 microg/mL, 6 days) or medium control. Two ANXA1-derived peptides, Ac2-26 and AF-2 (5-500 microM), were assessed for possible inhibition of PHA-and antigen-induced T cell proliferation (measured by 3H-thymidine uptake), while Ac2-26 was assessed for inhibition of Der p-induced interleukin (IL)-5 release and PPD-induced interferon-gamma (IFN-gamma) release (measured by ELISA). Comparison was made with dexamethasone as an established inhibitory control. Endogenous production by PBMC of cell surface-associated and intracellular ANXA1 in response to PHA, Der p and PPD in the presence and absence of dexamethasone was measured by specific ELISA.
RESULTS: Both PHA- and antigen-induced T cellular proliferation were inhibited by dexamethasone. Although neither ANXA1-derived peptide significantly altered PHA-induced proliferation, both effected concentration-dependent reductions in antigen-induced proliferation, Ac2-26 being the more potent. Peptides of identical amino acid composition to Ac2-26 and AF-2, but of random sequence, were ineffective at equivalent concentrations. In addition, Ac2-26 and dexamethasone inhibited Der p-induced IL-5 release and PPD-induced IFN-gamma release in a concentration-dependent fashion. Endogenous ANXA1 was detectable in PBMC, but at concentrations approximately 104-fold lower, in molar terms, than the effective concentrations of the exogenously added, ANXA1-derived inhibitory peptides. Endogenous production was not significantly altered by any of the T cell stimuli employed in this study, in the presence or absence of dexamethasone.
CONCLUSION: In prototype Th1 and Th2-type human T cell responses, ANXA1-derived peptides can inhibit antigen-driven cellular proliferation and cytokine production.
Kamal AM, Smith SF, De silva wijayasinghe M, Solito E, Corrigan CJ. Clin Exp Allergy. 2001;31(7):1116-25.
The appreciation that the inflammatory reaction does not ‘spontaneously’ finish, but rather that inflammatory resolution is an active phenomenon brought about by endogenous anti-inflammatory agonists opens multiple opportunities for a reassessment of the complexity of inflammation and its main mediators. This review dwells on one of these pathways, the one centered around the glucocorticoid-regulated protein Annexin A1 and its G protein-coupled receptor. In recent years, much of the knowledge detailing the processes by which Annexin A1 expresses its anti-inflammatory role on innate immunity has been produced. Moreover, the generation of the Annexin A1 null mouse colony has provided important proof-of-concept experiments demonstrating the inhibitory properties of this mediator in the context of inflammatory and/or tissue-injury models. Therefore, Annexin A1 acts as a pivotal homeostatic mediator, where if absent, inflammation would overshoot and be prolonged. This new understanding scientific information could guide us onto the exploitation of the biological properties of Annexin A1 and its receptor to instigate novel drug discovery programmes for anti-inflammatory therapeutics. This line of research relies on the assumption that anti-inflammatory drugs designed upon endogenous anti-inflammatory mediators would be burdened by a lower degree of secondary effects as these agonists would be mimicking specific pathways activated in our body for safe disposal of inflammation. We believe that the next few years will produce examples of such new drugs and the validity of this speculation could then be assessed.
Perretti M, Dalli J. Exploiting the Annexin A1 pathway for the development of novel anti-inflammatory therapeutics. Br J Pharmacol. 2009;158(4):936-46.
Myocardial reperfusion injury is associated with the infiltration of blood-borne polymorphonuclear leukocytes. We have previous described the protection afforded by annexin 1 (ANXA1) in an experimental model of rat myocardial ischemia-reperfusion (IR) injury. We examined the 1) amino acid region of ANXA1 that retained the protective effect in a model of rat heart IR; 2) changes in endogenous ANXA1 in relation to the IR induced damage and after pharmacological modulation; and 3) potential involvement of the formyl peptide receptor (FPR) in the protective action displayed by ANXA1 peptides. Administration of peptide Ac2-26 at 0, 30, and 60 min postreperfusion produced a significant protection against IR injury, and this was associated with reduced myeloperoxidase activity and IL-1beta levels in the infarcted heart. Western blotting and electron microscopy analyses showed that IR heart had increased ANXA1 expression in the injured tissue, associated mainly with the infiltrated leukocytes. Finally, an antagonist to the FPR receptor partially inhibited the protective action of peptide ANXA1 and its derived peptides against IR injury. Altogether, these data provide further insight into the protective effect of ANXA1 and its mimetics and a rationale for a clinical use for drugs developed from this line of research.
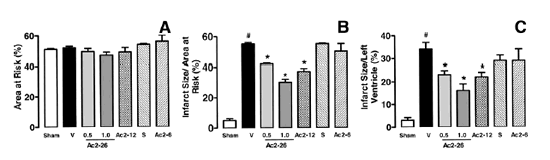
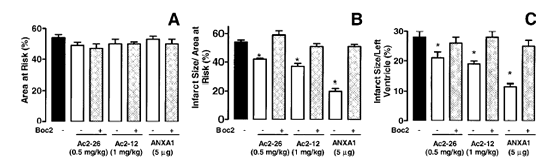
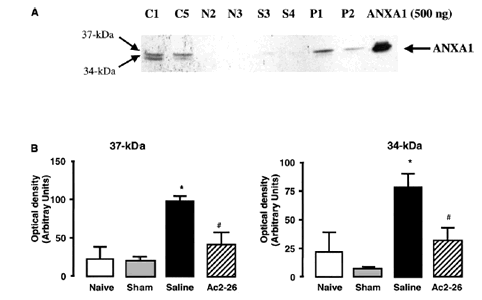
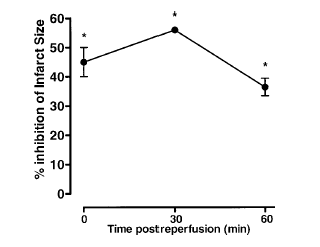
Effects of ANXA1-derived peptides on myocardial ischemia-reperfusion injury. Rats were treated i.v. with saline (1 ml/kg), peptide Ac2-26 (0.5 and 1 mg/kg), peptide Ac2-12 (1 mg/kg), scramble Ac2-12 (S, 1 mg/kg), or peptide Ac2-6 (1 mg/kg) at the end of the 25 min ischemic period. Tissues were analyzed 2 h after reperfusion; the area at risk (A), infarct size/area at risk (B), infarct size/left ventricle (C) were determined as described in Materials and Methods. A group of sham-operated animals (sham) was also evaluated. Data are means SE of n = 54 rats per group. #P < 0.05 vs. sham and *P < 0.05 vs. saline treatment. Boc2 reversed the cardioprotective actions of ANXA1 and its NH2 terminus peptides. The effect of peptide Ac2-26 (0.5 mg/kg) and ANXA1 (25 µg/kg) administered alone or together with 0.4 mg/kg Boc2. Drugs were administered 25 min after ischemia and tissues were analyzed 2 h after reperfusion; the area at risk (A), infarct size/area at risk (B), and infarct size/left ventricle (C) were determined as described in Materials and Methods. Data are means SE of n = 5 rats per group. *P < 0.05 vs. saline treatment. Expression of endogenous ANXA1 in myocardial samples. A) Western blot analysis of endogenous ANXA1 expression in sham (S), naive (N) animals, and those subjected to IR treated with either saline (C) or 1 mg/kg peptide Ac2-26 (P). B) Densitometric analysis for ANXA1 37 and 34 kDa isoforms. Values are mean SE of n = 5 rats per group. *P < 0.01 vs. sham treatment; #P < 0.01 vs. saline group. Time course of peptide Ac2-26 cardioprotective effect. Rats were given peptide Ac2?6 (1 mg/kg i.v.) immediately after the start of reperfusion (time 0), 30, and 60 min after reperfusion, and tissue was collected at 120 min to determine the infarct size/area at risk value as described in Materials and Methods. Infarct size was significantly reduced at all times vs. control (IS/AR value=55%, n=6). Data are mean SE of n = 4 rats per group. *P < 0.05 vs. control.
La M, D'amico M, Bandiera S, et al. FASEB J. 2001;15(12):2247-56.
High resolution kinetic data of the binding of fluorescent peptide to the N-formyl peptide receptor of neutrophils at 37°C has allowed for the development of a ligand binding model that predicts statistically larger binding rate constants than those previously reported for intact neutrophils. The new model accounts for ligand association and dissociation, receptor up-regulation, ligand-receptor complex internalization, a change in receptor affinity, and the quenching of internalized fluorescent ligand. We determined that receptor up-regulation is both agonist- and temperature-induced and is inhibited by both phenylarsine oxide and pertussis toxin treatment. Model fits of ligand association to pertussis toxin-treated cells show that while receptor up-regulation was inhibited, rate constants for ligand binding, receptor affinity conversion, and internalization of ligand-receptor complexes were unaffected. Results suggest Gi-protein-mediated receptor up-regulation and Gi-protein-independent receptor affinity conversion. Simulation of ligand infusion using our model gives insight into the quantitative and dynamic relationship between the low affinity ligand-receptor complex and the actin polymerization response. Hoffman JF, Linderman JJ, Omann GM. Receptor up-regulation, internalization, and interconverting receptor states. Critical components of a quantitative description of N-formyl peptide-receptor dynamics in the neutrophil. J Biol Chem. 1996;271(31):18394-404.
| Catalog# | Product | Standard Size | Price |
|---|---|---|---|
| 072-13 | Ac2-26 / Lipocortin-1 (2-26) / Annexin-1 (2-26) (Human) | 100 µg | $202 |
| 072-14 | Ac2-12 / Lipocortin 1 (2-12) / Annexin-1 (2-12) (Human) | 100 µg | $152 |
| FG-072-14A | Ac2-12 / Lipocortin 1 (2-12) / Annexin-1 (2-12) (Human) - FAM Labeled | 1 nmol | $317 |
| FR-072-14 | Ac2-12 / Lipocortin 1 (2-12) / Annexin-1 (2-12) (Human) - Rhodamine Labeled | 1 nmol | $317 |
| H-072-13 | Ac2-26 / Lipocortin-1 (2-26) / Annexin-1 (2-26) (Human) - Antibody | 50 µl | $571 |
| B-G-072-13 | Ac2-26 / Lipocortin-1 (2-26) / Annexin-1 (2-26) (Human) - Biotin Labeled Purified IgG | 100 µl | $635 |
| FC3-G-072-13 | Ac2-26 / Lipocortin-1 (2-26) / Annexin-1 (2-26) (Human) - Cy3 Labeled Purified IgG | 100 µl | $794 |
| FG-072-13A | Ac2-26 / Lipocortin-1 (2-26) / Annexin-1 (2-26) (Human) - FAM Labeled | 1 nmol | $382 |
| FG-G-072-13A | Ac2-26 / Lipocortin-1 (2-26) / Annexin-1 (2-26) (Human) - FAM Labeled Purified IgG | 100 µl | $635 |
| T-072-13 | Ac2-26 / Lipocortin-1 (2-26) / Annexin-1 (2-26) (Human) - I-125 Labeled | 10 µCi | $1082 |
Social Network Confirmation