
Abstract: Alzheimer's disease (AD) has been linked to multiple immune system-related genetic variants. Triggering receptor expressed on myeloid cells 2 (TREM2) genetic variants are risk factors for AD and other neurodegenerative diseases. In addition, soluble TREM2 (sTREM2) isoform is elevated in cerebrospinal fluid in the early stages of AD and is associated with slower cognitive decline in a disease stage-dependent manner. Multiple studies have reported an altered peripheral immune response in AD. However, less is known about the relationship between peripheral sTREM2 and an altered peripheral immune response in AD. The objective of this study was to explore the relationship between human plasma sTREM2 and inflammatory activity in AD. The hypothesis of this exploratory study was that sTREM2-related inflammatory activity differs by AD stage. We observed different patterns of inflammatory activity across AD stages that implicate early-stage alterations in peripheral sTREM2-related inflammatory activity in AD. Notably, fractalkine showed a significant relationship with sTREM2 across different analyses in the control groups that was lost in later AD-related stages with high levels in mild cognitive impairment. Although multiple other inflammatory factors either differed significantly between groups or were significantly correlated with sTREM2 within specific groups, three inflammatory factors (fibroblast growth factor-2, GM-CSF, and IL-1β) are notable because they exhibited both lower levels in AD, compared with mild cognitive impairment, and a change in the relationship with sTREM2. This evidence provides important support to the hypothesis that sTREM2-related inflammatory activity alterations are AD stage specific and provides critical information for therapeutic strategies focused on the immune response.
Weber GE, Khrestian M, Tuason ED, et al. Peripheral strem2-related inflammatory activity alterations in early-stage alzheimer’s disease. JI. 2022;208(10):2283-2299.
Background: Therapeutic modulation of TREM2-dependent microglial function might provide an additional strategy to slow the progression of Alzheimer's disease. Although studies in animal models suggest that TREM2 is protective against Alzheimer's pathology, its effect on tau pathology and its potential beneficial role in people with Alzheimer's disease is still unclear. Our aim was to study associations between the dynamics of soluble TREM2, as a biomarker of TREM2 signalling, and amyloid β (Aβ) deposition, tau-related pathology, neuroimaging markers, and cognitive decline, during the progression of autosomal dominant Alzheimer's disease.
Morenas-Rodríguez E, Li Y, Nuscher B, et al. Soluble TREM2 in CSF and its association with other biomarkers and cognition in autosomal-dominant Alzheimer’s disease: a longitudinal observational study. The Lancet Neurology. 2022;21(4):329-341.
Abstract: Triggering receptor expressed on myeloid cells 2 (TREM2) is an innate immune receptor expressed by macrophages and microglia in the central nervous system (CNS). TREM2 has attracted a lot of interest in the past decade for its critical role in modulating microglia functions under homeostatic conditions and in neurodegenerative diseases. Genetic variation in TREM2 is sufficient to cause Nasu-Hakola disease, a rare pre-senile dementia with bone cysts, and to increase risk for Alzheimer's disease, frontotemporal dementia, and other neurodegenerative disorders. Beyond the role played by TREM2 genetic variants in these diseases, TREM2 engagement is a key step in microglia activation in response to different types of tissue injury (e.g. β-Amyloid deposition, demyelination, apoptotic cell death) leading to enhanced microglia metabolism, phagocytosis, proliferation and survival. TREM2 also exists as a soluble form (sTREM2), generated from receptor shedding or alternative splicing, which is detectable in plasma and cerebrospinal fluid (CSF). Genetic variation, physiological conditions and disease status impact CSF sTREM2 levels. Clinical and preclinical studies suggest that targeting and/or monitoring sTREM2 could have clinical and therapeutic implications. Despite the critical role of sTREM2 in neurologic disease, its function remains poorly understood. Here, we review the current literature on sTREM2 regarding its origin, genetic variation, and possible functions as a biomarker in neurological disorders and as a potential active player in CNS diseases and target for therapies.
Filipello F, Goldsbury C, You SF, Locca A, Karch CM, Piccio L. Soluble TREM2: Innocent bystander or active player in neurological diseases? Neurobiology of Disease. 2022;165:105630.
Abstract: Triggering receptor expressed on myeloid cells-like 2 (TREML2) is a newly identified susceptibility gene for Alzheimer's disease (AD). It encodes a microglial inflammation-associated receptor. To date, the potential role of microglial TREML2 in neuroinflammation in the context of AD remains unclear. In this study, APP/PS1 mice were used to investigate the dynamic changes of TREML2 levels in brain during AD progression. In addition, lipopolysaccharide (LPS) stimulation of primary microglia as well as a lentivirus-mediated TREML2 overexpression and knockdown were employed to explore the role of TREML2 in neuroinflammation in the context of AD. Our results show that TREML2 levels gradually increased in the brains of APP/PS1 mice during disease progression. LPS stimulation of primary microglia led to the release of inflammatory cytokines including interleukin-1β, interleukin-6, and tumor necrosis factor-α in the culture medium. The LPS-induced microglial release of inflammatory cytokines was enhanced by TREML2 overexpression and was attenuated by TREML2 knockdown. LPS increased the levels of microglial M1-type polarization marker inducible nitric oxide synthase. This effect was enhanced by TREML2 overexpression and ameliorated by TREML2 knockdown. Furthermore, the levels of microglial M2-type polarization markers CD206 and ARG1 in the primary microglia were reduced by TREML2 overexpression and elevated by TREML2 knockdown. LPS stimulation increased the levels of NLRP3 in primary microglia. The LPS-induced increase in NLRP3 was further elevated by TREML2 overexpression and alleviated by TREML2 knockdown. In summary, this study provides the first evidence that TREML2 modulates inflammation by regulating microglial polarization and NLRP3 inflammasome activation. These findings reveal the mechanisms by which TREML2 regulates microglial inflammation and suggest that TREML2 inhibition may represent a novel therapeutic strategy for AD.
Wang SY, Fu XX, Duan R, et al. The Alzheimer’s disease-associated gene TREML2 modulates inflammation by regulating microglia polarization and NLRP3 inflammasome activation. Neural Regen Res. 2023;18(2):434.
Abstract: Variants in the triggering receptor expressed on myeloid cells 2 (TREM2) gene are associated with increased risk for late-onset AD. Genetic loss of or decreased TREM2 function impairs the microglial response to amyloid-β (Aβ) plaques, resulting in more diffuse Aβ plaques and increased peri-plaque neuritic dystrophy and AD-tau seeding. Thus, microglia and TREM2 are at a critical intersection of Aβ and tau pathologies in AD. Since genetically decreasing TREM2 function increases Aβ-induced tau seeding, we hypothesized that chronically increasing TREM2 signaling would decrease amyloid-induced tau-seeding and spreading. Using a mouse model of amyloidosis in which AD-tau is injected into the brain to induce Aβ-dependent tau seeding/spreading, we found that chronic administration of an activating TREM2 antibody increases peri-plaque microglial activation but surprisingly increases peri-plaque NP-tau pathology and neuritic dystrophy, without altering Aβ plaque burden. Our data suggest that sustained microglial activation through TREM2 that does not result in strong amyloid removal may exacerbate Aβ-induced tau pathology, which may have important clinical implications.
Jain N, Lewis CA, Ulrich JD, Holtzman DM. Chronic TREM2 activation exacerbates Aβ-associated tau seeding and spreading. Journal of Experimental Medicine. 2023;220(1):e20220654.
Abstract: Mutations in triggering receptor expressed on myeloid cells 2 (TREM2), which has been proposed to regulate the inflammatory responses and the clearance of apoptotic neurons and/or amyloid-β, are genetically linked to increased risk for late-onset Alzheimer's disease (AD). Interestingly, a missense variant in TREM-like transcript 2 (TREML2), a structurally similar protein encoded by the same gene cluster with TREM2 on chromosome 6, has been shown to protect against AD. However, the molecular mechanisms by which TREM2 and TREML2 regulate the pathogenesis of AD, and their functional relationship, if any, remain unclear. Here, we show that lipopolysaccharide (LPS) stimulation significantly suppressed TREM2 but increased TREML2 expression in mouse brain. Consistent with this in vivo result, LPS or oligomeric amyloid-β treatment down regulated TREM2 but up-regulated TREML2 expression in primary microglia. Most important, modulation of TREM2 or TREML2 levels had opposing effects on inflammatory responses with enhancement or suppression of LPS-induced proinflammatory cytokine gene expression observed on TREM2 or TREML2 down regulation, respectively. In addition, the proliferation of primary microglia was significantly decreased when TREM2 was down regulated, whereas it was increased on TREML2 knockdown. Together, our results suggest that several microglial functions are strictly regulated by TREM2 and TREML2, whose dysfunctions likely contribute to AD pathogenesis by impairing brain innate immunity. Our findings provide novel mechanistic insights into the functions of TREM2 and TREML2 in microglia and have implications on designing new therapeutic strategies to treat AD.
Zheng H, Liu CC, Atagi Y, et al. Opposing roles of the triggering receptor expressed on myeloid cells 2 and triggering receptor expressed on myeloid cells-like transcript 2 in microglia activation. Neurobiology of Aging. 2016;42:132-141.
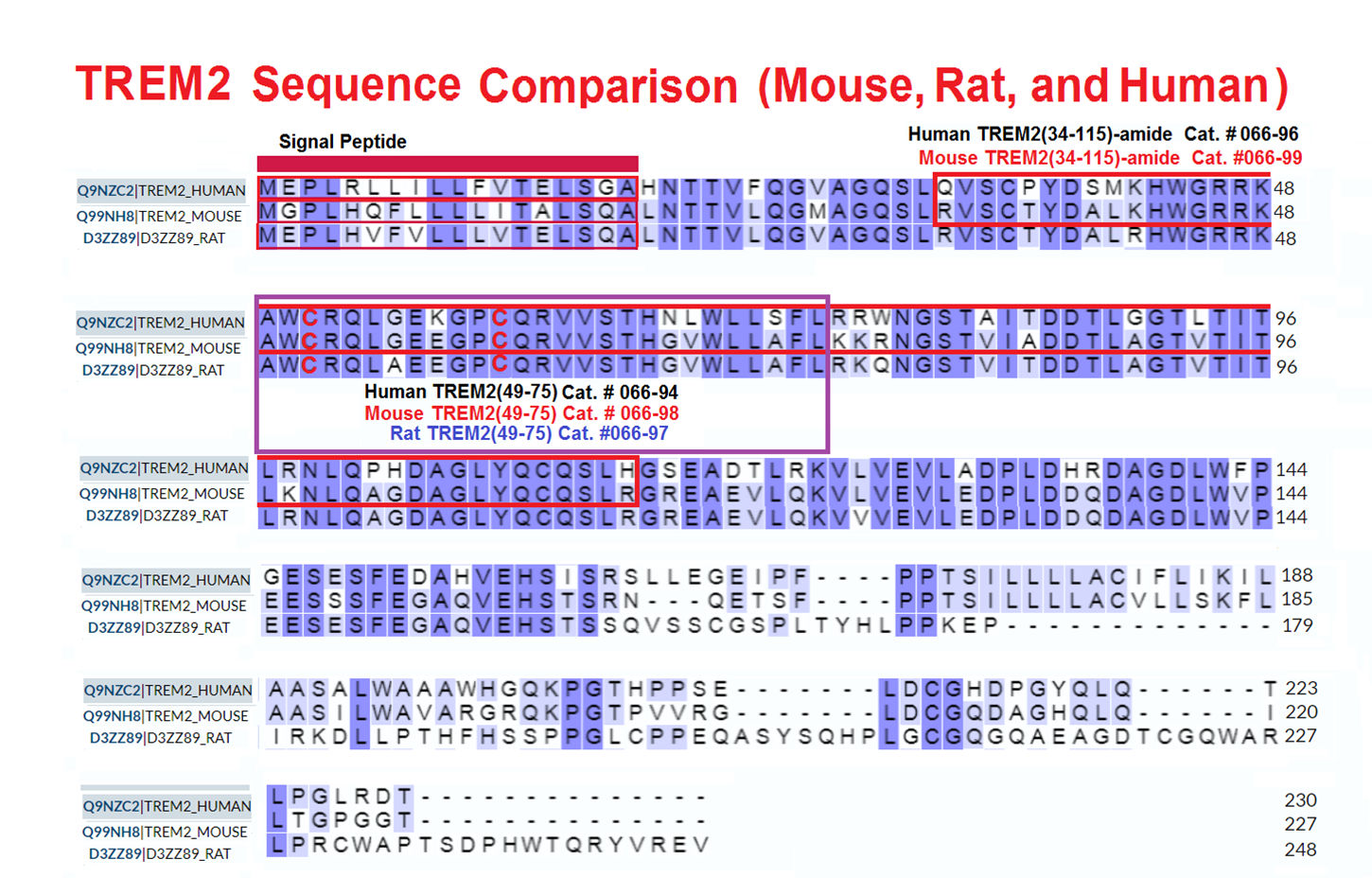
| Catalog# | Product | Standard Size | Price |
|---|---|---|---|
| 066-96 | TREM2 (34-115) amide (Human) | 100 µg | $425 |
| 066-99 | TREM2 (34-115) amide (Mouse) | 100 µg | $425 |
| 066-98 | TREM2 (49-75) (Mouse) | 100 µg | $187 |
| 066-97 | TREM2 (49-75) (Rat) | 100 µg | $187 |
| 066-94 | TREM2(49-75) (Human) | 100 µg | $187 |
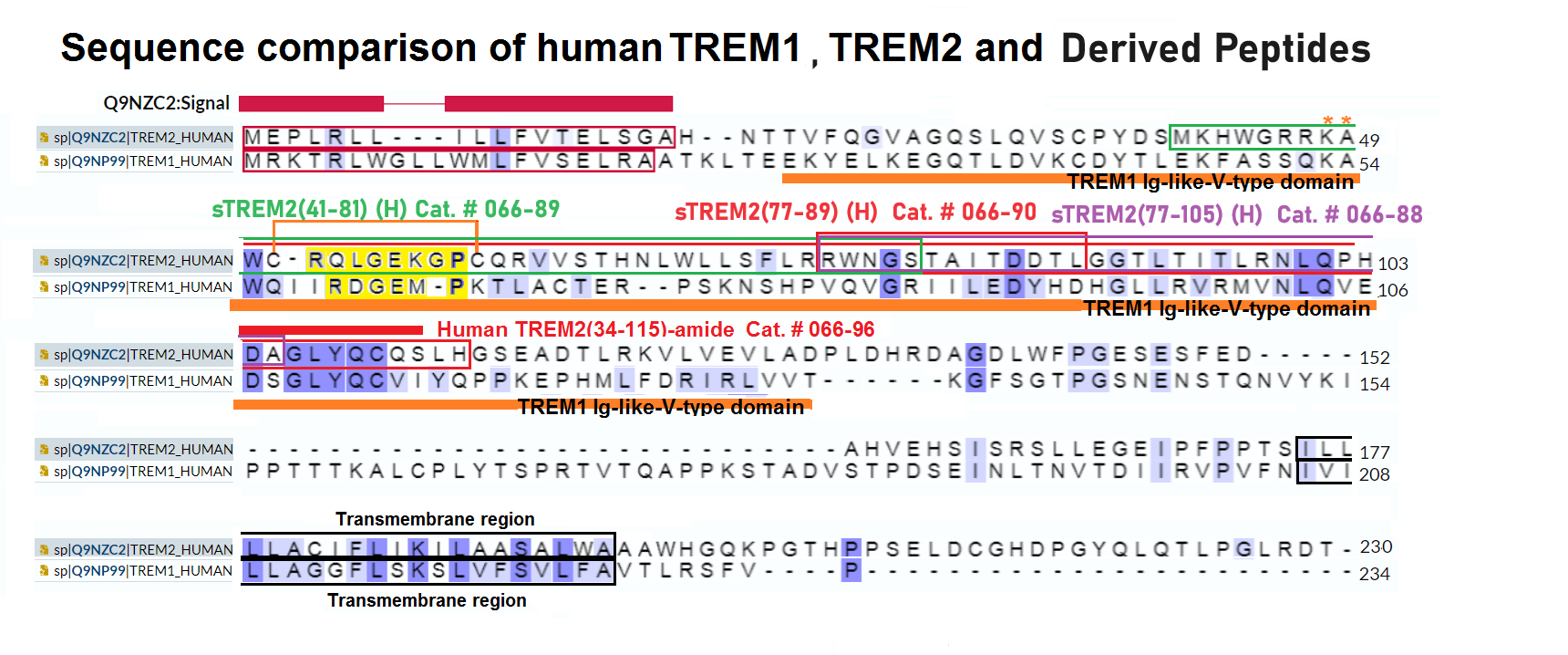
| 066-93 | TREML2 (39-107) amide (Human) | 100 µg | $425 |
| 066-91 | TREML2 (54-76) (Human) | 100 µg | $187 |
| 066-89 | sTREM2(41-81) (Human) | 100 μg | $247 |
| 066-88 | sTREM2(77-105) (Human) | 100 μg | $173 |
| 066-90 | sTREM2(77-89) (Human) | 200 μg | $131 |
Social Network Confirmation