|
|
 |
Alternative processing of the precursor protein pro-GIP results in endogenously produced GIP(1-30)NH2, that by DPP-4 cleavage in vivo results in the metabolite GIP(3-30)NH2. We showed previously that GIP(3-30)NH2 is a high affinity antagonist of the human GIPR in vitro. Here we determine whether it is suitable for studies of GIP physiology in rats since effects of GIP agonists and antagonists are strictly species-dependent. Transiently transfected COS-7 cells were assessed for cAMP accumulation upon ligand stimulation or assayed in competition binding using human 125I-GIP(1-42) as radioligand. In isolated perfused rat pancreata insulin, glucagon, and somatostatin-releasing properties were evaluated. Competition binding demonstrated that on the rat GIP receptor (GIPR), rat GIP(3-30)NH2 bound with high affinity (Ki of 17 nM), in contrast to human GIP(3-30)NH2 (Ki of 250 nM). In cAMP studies, rat GIP(3-30)NH2inhibited GIP(1-42)-induced rat GIPR activation and schild-plot analysis showed competitive antagonism with a pA2 of 13 nM and a slope of 0.9±0.09. Alone, rat GIP(3-30)NH2 displayed weak, low-potent partial agonistic properties (EC50>1µM) with an efficacy of 9.4% at 0.32 µM compared to GIP(1-42). In perfused rat pancreata, rat GIP(3-30)NH2 efficiently antagonized rat GIP(1-42)-induced insulin, somatostatin, and glucagon secretion. In summary, rat GIP(3-30)NH2 is a high affinity competitive GIPR antagonist and effectively antagonizes GIP-mediated G protein-signaling as well as pancreatic hormone release, while human GIP(3-30)NH2, despite a difference of only one amino acid between the two (arginine in position 18 in rat GIP(3-30)NH2; histidine in human), is unsuitable in the rat system. This underlines the importance of species differenes in the GIP system, and the limitations of testing human peptides in rodent systems.
Sparre-ulrich AH, Gabe MN, Gasbjerg LS, et al. Biochem Pharmacol. 2017;
BACKGROUND & AIMS: Glucose-dependent insulinotropic polypeptide (GIP) and the proglucagon product glucagon-like peptide-1 (GLP-1) are gastrointestinal hormones that are released in response to nutrient intake and promote insulin secretion. Interestingly, a subset of enteroendocrine cells express both GIP and GLP-1. We sought to determine whether GIP also might be co-expressed with proglucagon in pancreatic alpha-cells. METHODS: We assessed GIP expression via reverse-transcription polymerase chain reaction, in situ hybridization, and immunohistochemistry. We developed a novel bioassay to measure GIP release from isolated islets, compared the biological activities of full-length and truncated GIP, and assessed the impact of immunoneutralization of islet GIP on glucose-stimulated insulin secretion in isolated islets. RESULTS: GIP messenger RNA was present in mouse islets; GIP protein localized to islet alpha-cells of mouse, human, and snake pancreas, based on immunohistochemical analyses. However, using a C-terminal GIP antibody, immunoreactivity was detected in islets from prohormone convertase (PC) 2 knockout but not wild-type mice. Bioactive GIP was secreted from mouse and human islets after arginine stimulation. In the perfused mouse pancreas, GIP(1-42) and amidated GIP(1-30) had equipotent insulinotropic actions. Finally, immunoneutralization of GIP secreted by isolated islets decreased glucose-stimulated insulin secretion. CONCLUSIONS: GIP is expressed in and secreted from pancreatic islets; in alpha-cells, PC2 processes proGIP to yield a truncated but bioactive form of GIP that differs from the PC1/3-derived form from K-cells. Islet-derived GIP promotes islet glucose competence and also could support islet development and/or survival.
Fujita Y, Wideman RD, Asadi A, et al. Glucose-dependent insulinotropic polypeptide is expressed in pancreatic islet alpha-cells and promotes insulin secretion. Gastroenterology. 2010;138(5):1966-75
Glucose-dependent insulinotropic polypeptide (GIP) is a hormone released from enteroendocrine K cells in response to meals. Posttranslational processing of the precursor protein pro-GIP at residue 65 by proprotein convertase subtilisin/kexin type 1 (PC1/3) in gut K cells gives rise to the established 42-amino-acid form of GIP (GIP(1-42)). However, the pro-GIP peptide sequence contains a consensus cleavage site for PC2 at residues 52-55 and we identified PC2 immunoreactivity in a subset of K cells, suggesting the potential existence of a COOH-terminal truncated GIP isoform, GIP(1-30). Indeed a subset of mouse and human K cells display GIP immunoreactivity with GIP antibodies directed to the mid portion of the peptide, but not with a COOH-terminal-directed GIP antibody, indicative of the presence of a truncated form of GIP. This population of cells represents approximately 5-15% of the total GIP-immunoreactive cells in mice, depending on the region of intestine, and is virtually absent in mice lacking PC2. Amidated GIP(1-30) and GIP(1-42) have comparable potency at stimulating somatostatin release in the perfused mouse stomach. Therefore, GIP(1-30) represents a naturally occurring, biologically active form of GIP.
Fujita Y, Asadi A, Yang GK, Kwok YN, Kieffer TJ. Differential processing of pro-glucose-dependent insulinotropic polypeptide in gut. Am J Physiol Gastrointest Liver Physiol. 2010;298(5):G608-14
The hormonal factor(s) implicated as transmitters of signals from the gut to pancreatic-cells is referred to as incretin, and gastric inhibitory polypeptide (GIP) is identified as one of the incretins. GIP is a gastrointestinal peptide hormone of 42 amino acids that is released from duodenal endocrine K-cells after absorption of glucose or fat and exerts its effects by binding to its specific receptor, the GIP receptor. By generating and characterizing mice with a targeted mutation of the GIP receptor gene, we have shown that GIP has not only an insulinotropic role, but also physiological roles on fat accumulation into adipose tissues and calcium accumulation into bone. We here propose a new acronym, GIP, for gut-derived nutrient-intake polypeptide.
Yamada et al. Diabetes 55 (Suppl. 2):S86–S91, 2006
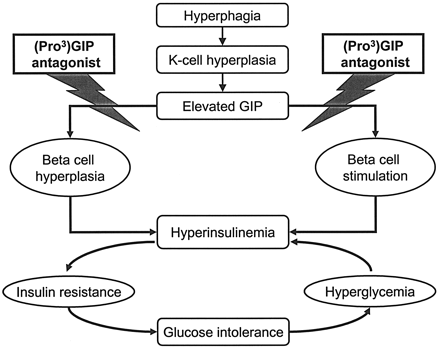
Glucose-dependent insulinotropic polypeptide (gastric inhibitory polypeptide [GIP]) is an important incretin hormone secreted by endocrine K-cells in response to nutrient ingestion. In this study, we investigated the effects of chemical ablation of GIP receptor (GIP-R) action on aspects of obesity-related diabetes using a stable and specific GIP-R antagonist, (Pro3)GIP. Young adult ob/ob mice received once-daily intraperitoneal injections of saline vehicle or (Pro3)GIP over an 11-day period. Nonfasting plasma glucose levels and the overall glycemic excursion (area under the curve) to a glucose load were significantly reduced (1.6-fold; P < 0.05) in (Pro3)GIP-treated mice compared with controls. GIP-R ablation also significantly lowered overall plasma glucose (1.4-fold; P < 0.05) and insulin (1.5-fold; P < 0.05) responses to feeding. These changes were associated with significantly enhanced (1.6-fold; P < 0.05) insulin sensitivity in the (Pro3)GIP-treated group. Daily injection of (Pro3)GIP reduced pancreatic insulin content (1.3-fold; P < 0.05) and partially corrected the obesity-related islet hypertrophy and beta-cell hyperplasia of ob/ob mice. These comprehensive beneficial effects of (Pro3)GIP were reversed 9 days after cessation of treatment and were independent of food intake and body weight, which were unchanged. These studies highlight a role for GIP in obesity-related glucose intolerance and emphasize the potential of specific GIP-R antagonists as a new class of drugs for the alleviation of insulin resistance and treatment of type 2 diabetes.
Gault VA, Irwin N, Green BD, et al. Chemical ablation of gastric inhibitory polypeptide receptor action by daily (Pro3)GIP administration improves glucose tolerance and ameliorates insulin resistance and abnormalities of islet structure in obesity-related diabetes. Diabetes. 2005;54(8):2436-46
| Catalog# | Product | Standard Size | Price |
|---|---|---|---|
| 027-10 | [D-Ala2]-GIP (Human) | 100 µg | $202 |
| 027-02 | GIP (Human) | 100 µg | $122 |
| 027-27 | GIP (Mouse) | 100 µg | $167 |
| EK-027-02 | GIP (Human) - EIA Kit | 96 wells | $570 |
| G-027-02 | GIP (Human) - Purified IgG Antibody | 400 µg | $414 |
| 027-29 | [Pro3]-GIP (Rat) | 100 µg | $202 |
| 027-49 | [Pro3]-GIP (Mouse) | 100 µg | $202 |
| 027-14 | [D-Ala2]-GIP (Rat) | 100 µg | $202 |
| 027-12 | GIP (Rat) | 100 µg | $122 |
| RK-027-02 | GIP (Human) - RIA Kit | 125 tubes | $880 |



Social Network Confirmation